
More Information
Submitted: 18 September 2019 | Approved: 03 October 2019 | Published: 04 October 2019
How to cite this article: Mohamed-Salem L, Rodríguez-Locarno TE, Moreno-Monsalve T, Castellón-Sánchez I, Contreras-Gutiérrez JF, et al. Scintigraphic non-invasive diagnosis of amyloid cardiomyopathy. J Cardiol Cardiovasc Med. 2019; 4: 156-158.
DOI: 10.29328/journal.jccm.1001058
ORCID ID: 0000-0002-1124-2553
Copyright License: © 2019 Mohamed-Salem L, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Computed tomography; DM; Coronary artery; Atherosclerosis

Scintigraphic non-invasive diagnosis of amyloid cardiomyopathy
Laroussi Mohamed-Salem*, Tomás E Rodríguez-Locarno, Tatiana Moreno-Monsalve, Isabel Castellón-Sánchez, José F Contreras-Gutiérrez and Antonia Claver-Valderas
Department of Nuclear Medicine, Virgen de la Arrixaca University Clinical Hospital, Murcia, Spain
*Address for Correspondence: Laroussi Mohamed-Salem, Department of Nuclear Medicine, Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain, Tel: 0034 616 87 96 19; Email: [email protected]
Amyloidosis encompasses a heterogeneous group of disorders, characterized by extracellular deposition of insoluble abnormal amyloid aggregates, due to a failure in protein quality control. Cardiac amyloidosis is a disorder in which proteins misfold and deposit as amyloid fibrils that infiltrate the myocardial extracellular space [1].
Transthyretin (ATTR) and light chain (AL) are the most frequent types of cardiac amyloidosis. Transthyretin is a protein mainly synthesized by the liver, it may be hereditary or acquired from either wild-type (ATTRwt) or mutant (ATTRm) amyloid [2]. Cardiomyopathy is a common manifestation of ATTR amyloidosis with a particularly poor life expectancy of 2 to 6 years after diagnosis [3]. Although considered rare, the prevalence of this serious disease is likely underestimated because symptoms can be non-specific, and diagnosis largely relies on amyloid detection in tissue biopsies.
We present the case of a 79-year-old male, former smoker with a history of heart failure, high blood pressure, chronic renal insufficiency and compensated chronic alcoholic liver disease. In 2010 presented an episode of heart failure with severe mitral regurgitation, functional deterioration and progressive heart failure symptoms, which required surgery in 2012, performing mitral repair, tricuspid ring placement and aortic biological valve implant, presenting post-operative paroxysmal atrial fibrillation. The patient’s medications included rivaroxaban, torasemide, nebivolol and candesartan. Several blood tests showed normal blood cell count and liver function, increased serum creatinine (2 mg/dl), as a result of chronic kidney failure and high plasma levels of NT-proBNP (16018 pg/ml) related to heart failure.
Transthoracic echocardiogram pointed out non-dilated left ventricle, with moderate to severe dysfunction, and severe hypertrophy, left ventricle ejection fraction of 55%, effective mitral plasty, normofunctional biological aortic prosthesis, right ventricular dilation with mild to moderate dysfunction and moderate to severe tricuspid insufficiency with severe pulmonary hypertension. Due to the high suspicion of amyloid infiltrative cardiomyopathy, 2-time abdominal fat biopsies were performed, that resulted negative for amyloid deposits. AL (light chain) amyloidosis was excluded by immunofixation electrophoresis of serum and urine and by the serum free light chain assay, demonstrating the absence of monoclonal protein in urine or plasma.
In order to identify the origin of amyloidosis, a Tc99m-DPD scan was requested, after obtaining written patient informed consent, a planar whole-body scintigraphy was performed 2 hours after intravenous injection of 740 MBq, that showed intense biventricular myocardial tracer uptake grade 3 (greater than the bone uptake), compatible with cardiac TTR amyloidosis, (Figures 1,2). Finally, a Genetic Testing resulted negative for TTR gene Mutation, which is consistent with the diagnosis of cardiac amyloidosis ATTRwt.
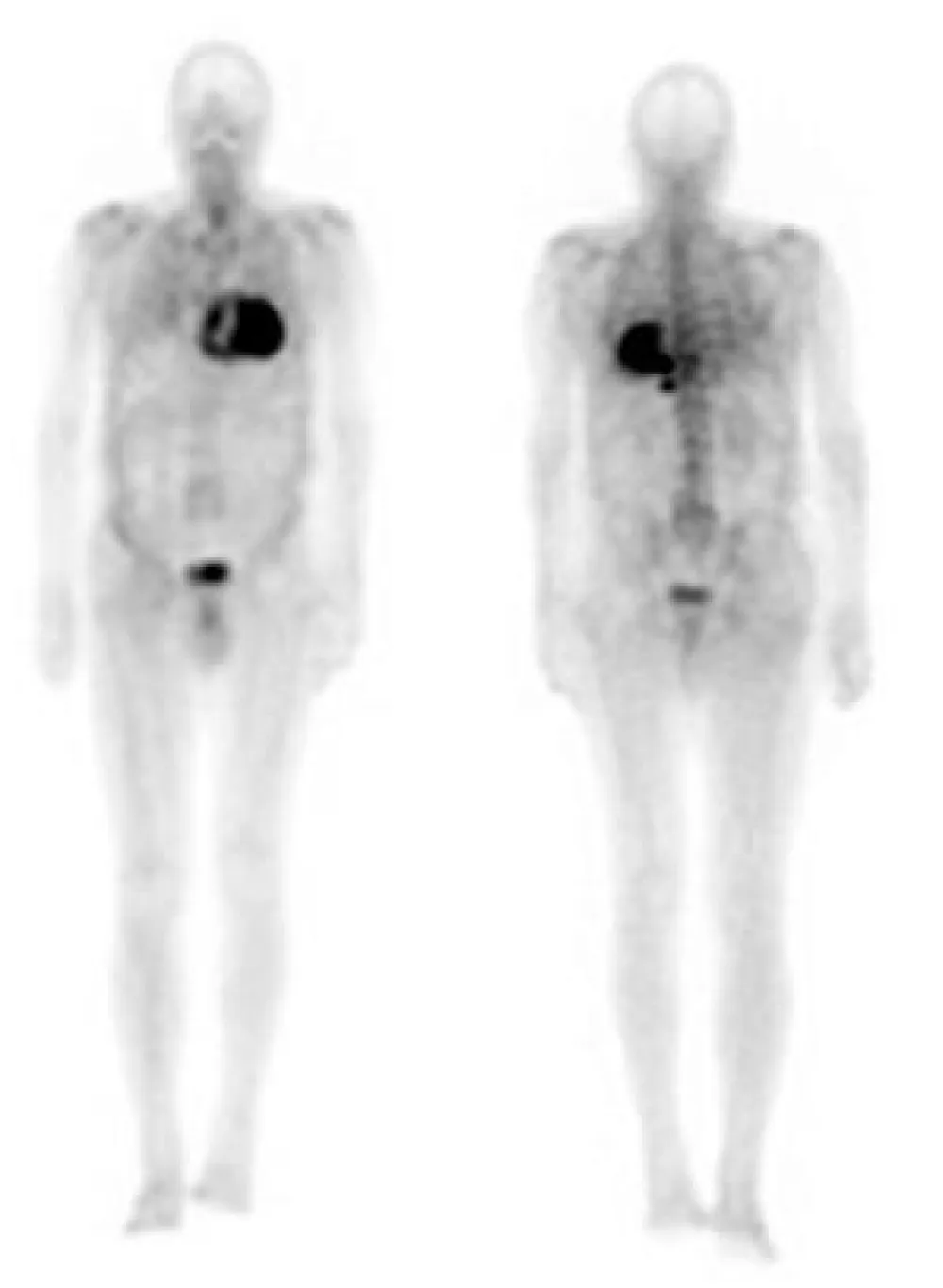
Figure 1: Anterior and posterior views whole body 99mTc-DPD scan. Marked myocardial uptake, suggesting an amyloid infiltration.
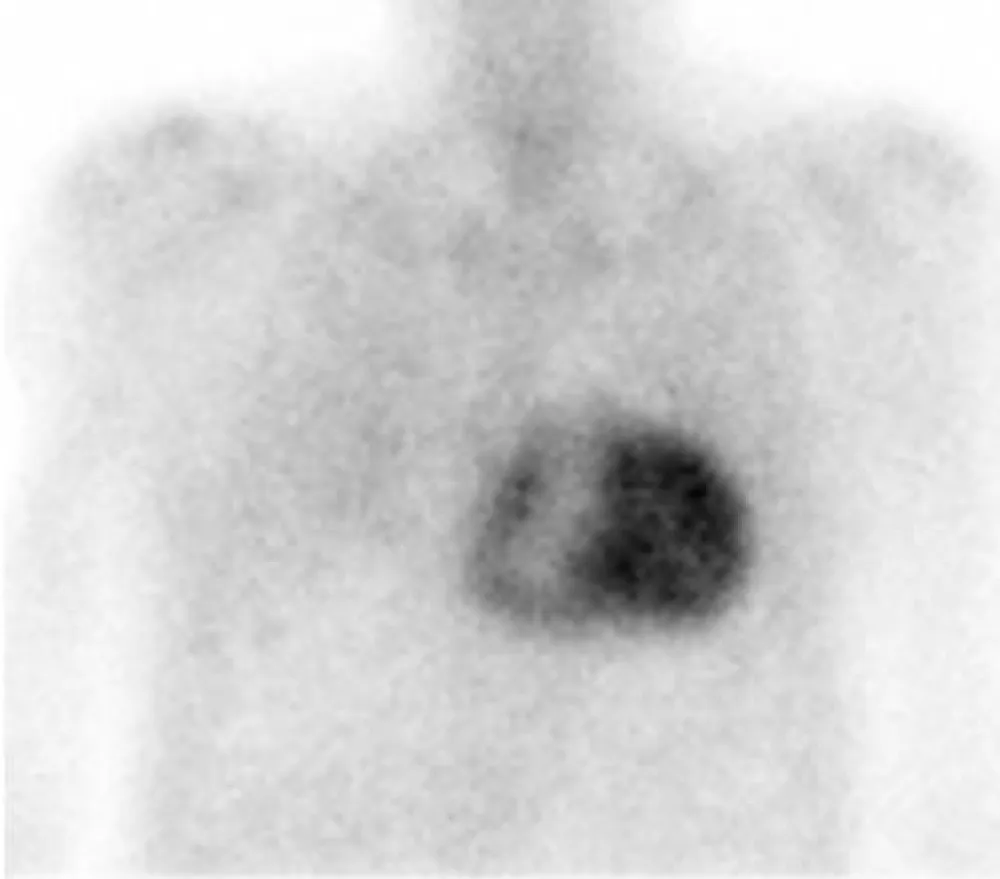
Figure 2: 99mTc-DPD anterior planar image show intense biventricular myocardial uptake (grade 3).
Cardiac transthyretin amyloidosis is a poorly recognized disease; the diagnosis is often delayed due to lack of knowledge of it. Cardiomyopathy or undifferentiated heart failure with preserved ejection fraction is a manifestation of this disease. Transthyretin protein misfolds to form fibrils that deposit in various tissues and organs. In addition to being debilitating, this disease is associated with a poor life expectancy, especially in those with cardiac dysfunction, however, the recent appearance of a wide range of effective therapeutic possibilities, allow early diagnosis and treatment is essential, since, the effectiveness of the treatment increases in the early stages of this disease [4].
ATTR may be hereditary due to variants in the TTR gene or acquired due to deposition of wild-type transthyretin (ATTRwt). The latter was formerly known as senile amyloidosis and almost exclusively affects the heart, causing myocardial infiltration and/or direct toxicity, leading to diastolic and systolic dysfunction, heart failure (HF), rhythm disturbances and ischemia [5].
It is very difficult to characterize each subtype of cardiac amyloidosis. Diagnostic confirmation of cardiac amyloidosis requires demonstration of amyloid deposit in a biopsy, although this does not necessarily have to be cardiac, if there are other organs other than the affected heart [6], however, it is the cardiac biopsy that ensures 100% sensitivity for disease detection [7]. But the invasive nature of endomyocardial biopsy and related high-risk complications are barriers, as is the specialist nature of immunohistochemistry and mass spectroscopy. Besides, as the lack of standardization on the definition of what constitutes cardiac amyloidosis, in some cases these is a need to use some ancillary imaging and clinical findings to ensure the pathological diagnosis. In addition, endomyocardial biopsy is classified in the class IIa recommendation [8].
The findings of a recent meta-analysis provide strong evidence of early use of nuclear scintigraphy among patients with a strong clinical suspicion of cardiac amyloidosis. Showing that nuclear scintigraphy works consistently in the diagnosis of cardiac amyloidosis and in the differentiation of its subtypes. It is important to note that additional investigations are required that combine scintigraphy with a triple detection of monoclonal proteins to reach the specificity close to 100% necessary for the diagnosis of ATTR cardiac amyloidosis [9].
The diagnosis of cardiac TTRwt amyloidosis is plausible in a patient with classical echocardiography characteristics of amyloid cardiomyopathy, and a technetium scan that shows a strong uptake associated with a negative genetic study, endomyocardial biopsy often is not necessary, particularly in the elderly or in patients debilitated by neuropathy. Noncardiac biopsies frequently will show amyloid deposits in patients with echocardiography or cardiac magnetic resonance findings consistent with amyloidosis, although positive noncardiac biopsies generally are less common in ATTR than in AL amyloidosis [10].
Bone tracer scintigraphy, using 99mTc-DPD, 99mTC-HMDP, and 99mTc-PYP, has been employed as a diagnostic test for ATTR. It is proven to be more than 99% sensitive for grades 1, 2, and 3 (the grading visual score, considers the myocardial tracer uptake compared with the bone uptake: grades 1 less than the bone uptake, grade 2 equal to the bone uptake and grade 3 greater than the bone uptake), and 97% specific for ATTR amyloid in the presence of grade 2 or 3 cardiac uptake [11]. It should be carried out under clinical suspicion for ATTR in the absence of monoclonal protein in the urine with a positive predictive value of 100% [11], as this case exhibits. Although the precise mechanism of DPD uptake in myocardium with amyloid deposits is not completely understood, it is likely due to the high calcium content in transthyretin amyloid fibrils [11].
In this case, the patient presented intense myocardial tracer uptake, together with the absence of monoclonal protein in urine or plasma and negative genetic test, which is consistent with ATTRwt cardiac amyloidosis. According to the latest scientific evidence, no endomyocardial biopsy is needed to confirm the diagnosis of this disease, which is now gaining well-deserved attention due to advances in imaging and the recent approval of targeted breakthrough therapies.
- Singh V, Falk R, Di Carli MF, Kijewski M, Rapezzi C, et al. State-of-the-art radionuclide imaging in cardiac transthyretin amyloidosis. J Nucl Cardiol. 2019; 26: 158-173. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30569412
- Falk RH. Diagnosis and management of the cardiac amyloidoses. Circulation. 2005; 112: 2047-2060. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16186440
- Patel KS, Hawkins PN. Cardiac amyloidosis: where are we today? J Intern Med. 2015; 278: 126-144. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26077367
- Maurer MS, Bokhari S, Damy T, Dorbala S, Drachman BM, et al. Expert Consensus Recommendations for the Suspicion and Diagnosis of Transthyretin Cardiac Amyloidosis. Circ Heart Fail. 2019; 12: e006075. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/31480867
- Mohamed-Salem L, Santos-Mateo JJ, Sanchez-Serna J, Hernández-Vicente Á, Reyes-Marle R, et al. Prevalence of wild type ATTR assessed as myocardial uptake in bone scan in the elderly population. Int J Cardiol. 2017; 270: 192-196. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/29903517
- Mohamed-Salem L, Moreno-Monsalve T, Castellón-Sánchez MI, Claver-Valderas MA, Pascual-Figal DA. Non-biopsy diagnosis of familial amyloid cardiomyopathy. J Nucl Cardiol. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30945209
- Haro-del F, Sánchez A, Gómez M, García P, Salas-Antón C, et al. Role of cardiac scintigraphy with 99mTC-DPD in the discrimination of the cardiac amyloidosis subtype. Rev Esp Cardiol. 2012; 65: 440-446.
- Zhao L, Tian Z, Fang Q. Diagnostic accuracy of cardiovascular magnetic resonance for patients with suspected cardiac amyloidosis: a systematic review and meta-analysis. BMC Cardiovascular Disorders. 2016; 16: 12. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27267362
- Brownrigg J, Lorenzini M, Lumley M, Elliott P. Diagnostic performance of imaging investigations in detecting and differentiating cardiac amyloidosis: a systematic review and meta-analysis. ESC Heart Failure. 2019.
- Ertz MA, Benson MD, Dyck PJ, Grogan M, Coelho T, et al. Diagnosis, prognosis, and therapy of transthyretin amyloidosis. J Am Coll Cardiol. 2015; 66: 2451-2466. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26610878
- Gillmore JD, Maurer MS, Falk RH, Merlini G, Damy T, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016; 133: 2404-2412. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27143678