
More Information
Submitted: 29 February 2020 | Approved: 18 April 2020 | Published: 20 April 2020
How to cite this article: Uca YO, Taupitz M. Glycosaminoglycans as Novel Targets for in vivo Contrast-Enhanced Magnetic Resonance Imaging of Atherosclerosis J Cardiol Cardiovasc Med. 2020; 5: 080-088.
DOI: 10.29328/journal.jccm.1001091
ORCiD: orcid.org/0000-0003-3268-4382
Copyright License: © 2020 Uca YO, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Glycosaminoglycans as Novel Targets for in vivo Contrast-Enhanced Magnetic Resonance Imaging of Atherosclerosis
Yavuz O Uca* and Matthias Taupitz
Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt Universität zu Berlin, and Berlin Institute of Health, Charité Center Diagnostic and Interventional Radiology and Nuclear Medicine 10117, Berlin, Germany
*Address for Correspondence: Yavuz Oguz Uca, Charité - Universitätsmedizin Berlin, corporate member of Freie Universität Berlin, Humboldt Universität zu Berlin, and Berlin Institute of Health, Charité Center Diagnostic and Interventional Radiology and Nuclear Medicine, Charitéplatz 1, 10117 Berlin, Germany, Tel: +49 (30) 450539066; Email: [email protected]
Atherosclerosis is an important promoter of cardiovascular disease potentiating myocardial infarction or stroke. Current demand in biomedical imaging necessitates noninvasive characterization of arterial changes responsible for transition of stable plaque into rupture-prone vulnerable plaque. in vivo contrast enhanced magnetic resonance (MR) imaging (MRI) allows quantitative and functional monitoring of pathomorphological changes through signal differences induced by the contrast agent uptake in the diseased vessel wall, therefore it is the ideal modality toward this goal. However, studies have so far focused on the cellular targets of persisting inflammation, leaving extracellular matrix (ECM) far behind. In this review, we portray ECM remodeling during atherosclerotic plaque progression by summarizing the state of the-art in MRI and current imaging targets. Finally, we aim to discuss glycosaminoglycans (GAGs) and their functional interactions, which might offer potential toward development of novel imaging probes for in vivo contrast-enhanced MRI of atherosclerosis.
Atherosclerosis is a systemic inflammatory disorder which affects majorly large and medium-sized arteries [1]. It is a promoter of cardiovascular disease (CVD) potentiating myocardial infarction or stroke [2]. Molecular imaging studies utilizing different modalities have resulted in better understanding of the disease mechanisms, however the gravity of the health and economic burden of CVD calls for improvements in the diagnosis [3-5]. Current demand in biomedical imaging entails in vivo detection of atherosclerotic plaques with quantitative or predictive value through identifying vessel wall alterations leading to rupture-prone vulnerable plaque [6].
Contrast-enhanced magnetic resonance (MR) imaging (MRI) is the ideal modality towards this goal [7]. It allows non-invasive discrimination of major pathomorphological changes in atherosclerotic plaque progression: lipid core, fibrous cap, calcifications, intraplaque hemorrhage and acute thrombosis [8]. The use of gadolinium (Gd)‑based contrast agents (CAs) on the clinical level and iron oxide nanoparticles (IONPs) in experimental studies have placed plaque biology ahead of luminal stenosis [8-12]. Coexisting with the lipid accumulation and retention, inflammatory mediators are currently extensively investigated by MRI, in which cellular components are the most attractive targets [13-17].
Although generally ignored in the field of molecular imaging, considerable attention has recently been raised toward molecular imaging of the extracellular matrix (ECM) in the context of atherosclerosis [18]. Apart from being the major component of the atherosclerotic plaque ultrastructure, coordinate synthesis and turnover of the ECM is a characteristic feature of plaque instability [19]. ECM alterations during arterial remodeling in atherosclerotic plaque progression involve highly abundant molecular interactions, which exhibit importance toward unraveling new targets, thus developing novel imaging probes.
Among ECM components, glycosaminoglycans (GAGs) are well known for their antigenic characteristics in drug development or tissue engineering [20,21]. Following reports on tissue deposition of Gd in nephrogenic systemic fibrosis (NSF), a fatal disease observed in patients with end stage renal disease who had received Gd-based MR CAs, studies have demonstrated colocalization of elements including phosphorus or calcium [22,23]. Insoluble complex formation through transchelation of Gd by physiological anions such as glycosaminoglycans has been postulated as a plausible underlying mechanism [24,25].
In this review, we aim to explore potentials of GAGs for in vivo contrast-enhanced MR imaging of atherosclerosis with a deeper look into their features as information-loaded sensing molecules of the ECM. We will try to elucidate ECM remodeling during atherosclerotic plaque progression by examining distributions and dynamic alterations of GAGs. Finally, we will discuss functional characteristics of GAGs in molecular interactions, which could guide investigations aiming at developing novel targets to identify vessel wall alterations leading to rupture-prone vulnerable plaque.
Atherogenesis and imaging targets
Atherogenesis is the interplay of smooth muscle cells (SMCs), immune cells, fibroblasts and the ECM (Figure 1) [26]. The native endothelium maintains vascular homeostasis by the support of laminar shear stress within the arteries [27]. Altered flow conditions where turbulent flow dominates over laminar sheer stress transform the endothelium into permeable and leaky form resulting in its activation [28]. This accounts for early atherogenesis, which is characterized by the formation of neointima through low density lipoprotein (LDL) influx and subsequent oxidative modification [29]. Endothelial cell adhesion molecules (CAMs), transmigration of monocytes or lymphocytes through interactions with selectins or integrins and angiogenesis offer a range of imaging targets [30-32]. Among these, vascular cell adhesion molecule-1 (VCAM-1) and αvβ3-integrin have been extensively investigated [33,34]. Additionally, proinflammatory cytokines or chemokines including interleukin-1 (IL-1), monocyte chemotactic protein-1 (MCP-1) and tumor necrosis factor-α (TNF‑α) are attractive imaging targets of early atherogenesis [35,36].
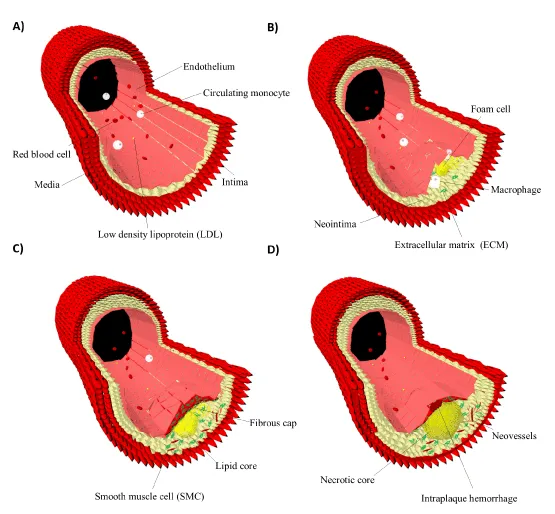
Figure 1: Atherosclerotic plaque progression. The native endothelium maintains vascular homeostasis (A). Early atherogenesis is characterized by the formation of the neointima through lipid influx, monocyte transmigration and foam cells (B). Persisting inflammation during atherosclerotic plaque progression leads to formation of lipid cores. Arterial remodeling results in increased vasa vasorum activity forming neovessels. In parallel, medial smooth muscle cells undergo phenotypic changes and migrate into the neointima. Here, they proliferate and produce new extracellular matrix molecules forming the fibrous cap. Tissue breakdown and thinning of the fibrous cap provoke erosion and renders the plaque vulnerable by inducing tears, intraplaque hemorrhage and thrombosis (C,D) [19,38].
Arterial remodeling and increased vasa vasorum activity during plaque progression substantiate expression of early markers and expose diversity in target molecules [1-,37]. LDL influx escalates transmigration of monocytes into the neointima, where they differentiate into mature macrophages. Macrophages are the major inflammatory elements of both early and late atherosclerotic lesions, thereby their phagocytic activity has so far been the most attractive target for contrast-enhanced MRI [38,39]. Phagocytosis offers a unique strategy as it results in concentration of CAs. Macrophage uptake of IONPs in atherosclerotic plaques was reported for the first time in a rabbit model, which was followed by a multicenter phase III clinical trial documenting accumulation of IONPs around the inflamed regions of atherosclerotic plaques [40,41]. Shortly after, in an in vivo study on human ruptured and rupture-prone lesions, Kooi, et. al. reported substantial signal decrease 24 h after intravenous administration [42].
Besides phagocytosis, proteolysis as a cohort of SMCs, macrophages, mast cells and T-lymphocytes allows monitoring of ECM degradation and thinning of the collagen-rich fibrous cap [43,44]. Matrix metalloproteinases (MMPs), cathepsin or serine proteases, chymase, tryptase or stromelysin-1 are targeted to assess the susceptibility to rupture or thrombosis [45-47]. Finally, screening for upregulated vasa vasarum activity, late stage immune cell antigens, activated platelets, fibrin and apoptosis markers such as annexin-V are among major targets of imaging vulnerable plaques [48-50].
MR imaging of the extracellular matrix
ECM is a highly organized three-dimensional network of fiber forming and non-fiber forming molecules cross-linked into a biomechanically viscoelastic composition (Figure 2) [51]. ECM components consist of collagen, elastin and fibrin, proteoglycans (PGs), glycosaminoglycans (GAGs) and glycoproteins such as fibronectin, vitronectin, laminin and tenascin [20,52]. Deposition and remodeling of the ECM are among the hallmarks of atherosclerosis [53]. Early atherosclerotic lesions are characterized by the deposition of molecules that create a loose matrix, also known as ‘provisional matrix’ [54]. The major molecular composition of this proliferative phase consists of fibronectin, tenascin and thrombospondin [55]. ECM of fatty streaks in human atheroma was shown to contain substantial level of fibronectin, which attracts fibrillogenesis and fibronectin isoforms for MR imaging of early angiogenesis [56,57].
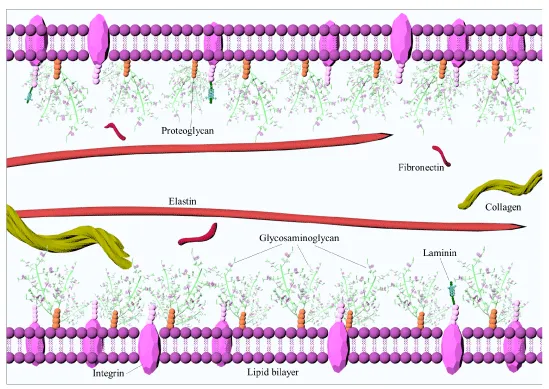
Figure 2: Extracellular matrix structure. Extracellular matrix is a highly organized three dimensional network of fiber forming and non-fiber forming molecules cross-linked into a biomechanically viscoelastic composition [51]. It’s major components are collagen, elastin, fibrin, proteoglycans, glycosaminoglycans, glycoproteins such as fibronectin, vitronectin, laminin and tenascin.
Transformation of the provisional matrix to fibrous matrix happens upon upregulation of the fibrous ECM components [55]. Collagen is the principal constituent of this state with varying types and tissue distributions [58]. It’s relative abundance, major role in maintaining the structural integrity and participation in the key inflammatory reactions make it a valuable target [59-61]. Collagenous thickening of the fibrous cap, retention of oxidized LDL (ox-LDL) by binding to collagen type I, III, IV and V as well as its turnover by the MMP activity result in a large pool of imaging targets [62-66].
Elastin is the second most abundant fibrous constituent of the ECM, majorly found in the media of the healthy arterial wall [67]. Besides maintaining the structural integrity, it prevents LDL penetration by forming lamellar layers [68]. Degradation and fragmentation of elastin, thus its absence in the newly remodeled matrix lead to increased LDL retention [69]. Alterations between elastogenesis and the turnover empower it’s use in monitoring lesion progression and determining the propensity into the rupture‑prone plaque [70,71]. Characterization and quantification of the plaque burden by an elastin‑targeting CA was reported promising for screening large human populations [72,73].
Fibrin, although less abundant then collagen and elastin, is another fibrous molecule especially overexpressed in the ECM of advanced plaques with erosions, which result in fissures reaching into the necrotic core and hemorrhage [74]. Fibrin-rich ECM aggregation leads to activation of the coagulation cascades, and recruitment of proinflammatory cells [75,76]. Domain-based modular interactions and the procoagulant activity render fibrin an important target for the detection of high-risk plaques with subacute and acute indications [77-80].
Glycosaminoglycans
GAGs are linear polysaccharides consisting of alternating disaccharide units of an amino sugar and uronic acid mostly found covalently attached to a core protein (Figure 3) [81]. They are a minor constituent of the healthy arterial wall, but their upregulation during lesion progression has been well documented [82,83]. Important types of GAGs in the vasculature are heparan sulfate (HS), heparin, chondroitin sulfate (CS), dermatan sulfate (DS) and hyaluronan (HA) [84]. GAGs differentiate from one another by their monomeric building blocks, position and configuration of the glycosidic linkages, chain length, and by the degree and position of sulfation and epimerization [85]. All GAGs except HA are sulfated or epimerized at variable degrees, which are coordinated in a specific manner [85]. The combinatorial biosynthetic process, chain elongation, and modifications give rise to an incredible structural diversity, imposing GAGs to be the most information-dense molecules in biology [86,87].
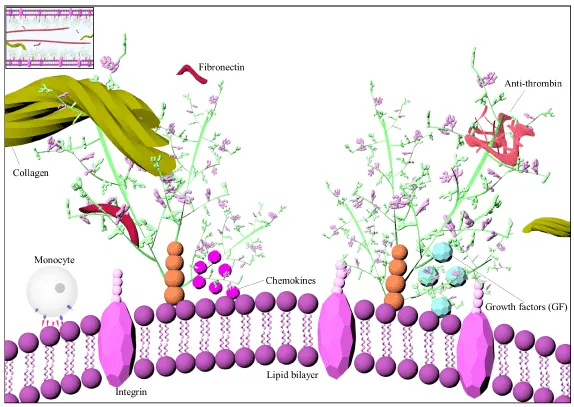
Figure 3: Illustration of glycosaminoglycan structure and interactions. Glycosaminoglycans are linear polysaccharides consisting of alternating disaccharide units of an amino sugar and uronic acid mostly found covalently attached to a core protein [81]. Glycosaminoglycan chains contribute to tertiary complex formation through establishing electrostatic bridges between nearby molecules and assist higher level interactions involving hydrophobic forces, hydrogen bonding or ionic interactions between proteins [99,100].
Immunohistochemical studies on atherosclerotic lesion progression have revealed distinct topography of GAGs with differences in spatial and temporal distribution [88]. Overall, CS/DS is known to increase, and HS is known to decrease [89-91]. Early lesions with stable endothelium, contractile SMC phenotype and occasional macrophage content display GAG distribution restricted to the cellular areas, whereas advanced lesions characterized by dysfunctional endothelium, synthetic SMC phenotype and macrophage content display highly complex distribution [88,92,93].
GAGs play important roles in supporting the structural organization of the ECM, regulating viscoelasticity and tissue permeability, lipid metabolism, cell signaling, migration, and differentiation [94-96]. Some of their critical functions have been decoded through genetic studies, in which manipulations of genes encoding their biosynthetic enzymes resulted with severe disruptions [97]. GAGs exert their functions through localization, stabilization, activation or inactivation of proteins (Figures 3,4) [98]. GAG chains contribute to complex formation through establishing electrostatic bridges and assist higher level interactions [99,100]. For instance, GAG‑LDL complexes were reported to be internalized by macrophages easier than LDL alone [101]. Lipid retention happens upon initial electrostatic binding with CS/DS chains of the PGs secreted by vascular SMCs and is followed by hydrophobic self-association of the lipoprotein [102,103]. However, positively charged amino acids on the apolipoprotein B, and their interactions with the sulfate or carboxylate groups on the GAG chains are crucial for binding [104,105].
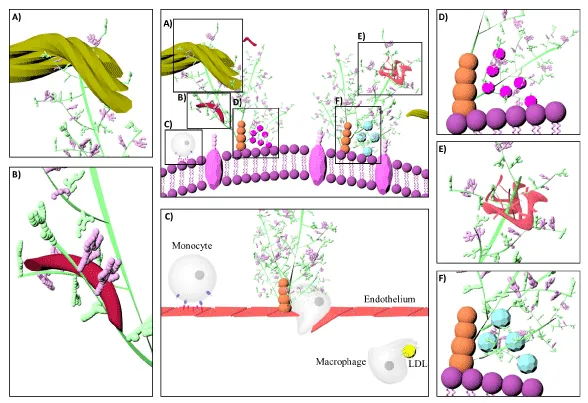
Figure 4: Schematic illustration of different glycosaminoglycan interactions. Glycosaminoglycans localize, stabilize, activate or inactivate proteins [98]. Decorin is a proteoglycan form of dermatan sulfate, which binds to collagen (A). Heparin interacts with fibronectin (B) [133]. Monocyte transmigration happen through interactions with the cell surface heparan sulfates on the endothelium and glycosaminoglycan low density lipoprotein complexes are more easily internalized by macrophages than lipoproteins alone (C) [101,127]. Glycosaminoglycans serve as storage reservoir for chemokines (D) and growth factors (F) [128]. They perform the highest affinity binding by inducing conformational changes and surface complementarity on the proteins [111,112]. Heparan sulfate-fibroblast growth factor and heparin anti thrombin III are the most well-known examples for such interactions (E).
Versatile binding ability permits different GAGs to interact with the same protein or different proteins simultaneously (Figure 4) [85,106]. Platelet factor-4 (PF4) binding is good example for cooperative activity [107]. During coagulation, PF4 is released from the α-granules of activated platelets by the coordinated transfer from the CS to more sulfated polysaccharides [108]. PF4 binds to and neutralizes heparin upon which it modulates fibroblast growth factor-2 (FGF-2) activity [109]. Similarly, the interaction between PF4 and neutrophil cell surface CS constitutes an essential mechanism for modulation of immune response by GAGs serving as physiologically relevant receptors [110].
Finally, GAGs perform the highest affinity binding by inducing conformational changes and surface complementarity on the proteins, attributed to torsional angle changes in the backbone chains of GAGs [111,112]. HS-FGF and heparin‑anti thrombin‑III (AT‑III) are the most well-known examples for such interactions. HS enhances FGF binding and oligomerization at its receptor owing to the presence of rare sulfation and epimerization patterns on at least tetrasaccharides [113,114]. AT-III is an inhibitor in the coagulation cascade, which binds to thrombin in 1:1 molar ratio [81]. Heparin forms a tight ternary complex with the AT-III through a specific pentasaccharide consensus motif and accelerates the rate of thrombin inhibition [111,115]. A unique sequence having four anionic groups (two N- and two O-sulfates, 3‑O-sulfation being critical) on the glucosamine residues has been shown to be crucial for the ionic contacts with the protein [116]. DS‑heparin cofactor II interaction was also reported to take place through binding of a specific DS hexasaccharide, which constitutes only 2% of hexasaccharides in DS, rendering it an important example for rare modifications on GAGs necessary for their functional roles [117,118].
GAGs as novel targets in contrast-enhanced MRI
Toward the goal of detecting atherosclerotic plaques at early stages, it is imperative to enhance the diagnostic value of existing approaches in characterizing the transition of stable plaque into vulnerable plaque [16]. In that regard, high spatial resolution-based investigations determining microdistribution of CAs in atherosclerotic plaques and their interactions with the ECM components would be beneficial [119]. Additionally, immunohistochemical investigations supporting existing MRI approaches should be reinforced by GAG immunostaining methods and complemented by elemental microscopy [120-122]. GAGs are a minor constituent of the healthy arterial wall but their rapid upregulation during lesion formation and progression along with their functional roles in major inflammatory processes highlight their potency in developing novel CAs that might offer advantages over existing MRI methods [82,83].
In summary, the contrast function of the agents administered arise from catalytically shortening the relaxation times of bulk water protons [123]. The polyelectrolyte nature of GAGs exposes them as the major charge hotspots in the ECM, making it suitable for exploring their properties in binding to and clustering CAs [124]. Also, lesion formation induces high expression of antigenic structures. These are almost always in the form of GAGs or glycoprotein conjugates [20]. Immune cells are often activated by these molecules and guided to the site of action through interactions of chemokines or cytokines with GAGs [125,126]. This applies to concentration of growth factors (GFs), enzymes or enzyme inhibitors as well [127,128]. Identifying types or compositions of GAG molecules in such interactions allow the mimicry of these by GAG‑CA conjugates. Similarly, major pathomorphological changes during lesion progression involve GAGs, which can be tailored to purpose of imaging. Among those, interactions of CS/DS chains secreted by vascular SMCs with LDL molecules and hydrophobic self-association might serve well for the development of novel liposome-based CAs [69]. Moreover, decorin is a PG-form of DS that binds to collagen, thereby might be engineered to monitor processes including thinning of the fibrous cap, erosions or calcifications [93,129]. Notably, elastin assembly, elastin binding of fibronectin or coagulation cascades, which display versatile affinity binding ability of GAGs offer useful strategies for targeting approaches not only through traditional protein components, but also through GAGs [85,106,130].
Finally, sulfation holds the key for elucidating unfamiliar features of GAGs during atherosclerotic plaque progression [131]. Variations in type, position or degree of sulfation, which result in distinct domain formations with particular or even rare-type of modifications are the core factors underlying their functional interactions [132]. Thus, it is highly conceivable that better understanding of these features is promising toward unraveling new targets, and development of more sophisticated MR contrast agents.
In this review, we aimed to explore potentials of GAGs for vivo contrast-enhanced MR imaging of the ECM in the context of atherosclerosis. Diseases present qualitative and quantitative changes in the ECM, in which GAGs take on diverse functional roles. ECM has been receiving attention as a potential area of research in molecular imaging. In parallel, technological advances in glycobiology have been increasing, among which chemical synthesis of oligosaccharides with desired modifications holds great potential for future investigations. Differential spatial or temporal distribution of GAGs in the vasculature, their rapid upregulation upon atherosclerotic plaque formation and functional roles in molecular interactions arising from versatile binding characteristics praise them as potent candidates toward development of novel imaging probes for in vivo contrast-enhanced MRI of atherosclerosis.
We thank Dr. Jörg Schnorr, Dr. Antje Ludwig, Dr. Judith Bergs, Prof. Dr. Tobias Schäffter, Prof. Dr. Ingolf Sack for their valuable support and Rafet Güven for graphical artwork.
- Ross R. Atherosclerosis — An Inflammatory Disease. N Engl J Med. 1999; 340: 115–126. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9887164
- Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, et al. Heart disease and stroke statistics-2019 update: a report From the American Heart Association. Circulation. 2019; 139 :526–528. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/30700139
- Nichols M, Townsend N, Scarborough P, Rayner M. Cardiovascular disease in Europe 2014: epidemiological update. Eur Heart J. 2014; 35: 2950–2959. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/25139896
- Heidenreich PA, Trogdon JG, Khavjou OA, Butler J, Dracup K, et al. Forecasting the Future of Cardiovascular Disease in the United States: A Policy Statement From the American Heart Association. Circulation. 2011; 123: 933–944. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21262990
- Nojiri S, Daida H. Atherosclerotic Cardiovascular Risk in Japan. Jpn Clin Med [Internet]. 2017; 8. PubMed: http://www.ncbi.nlm.nih.gov/pmc/articles/PMC5480958/
- Pozo E, Agudo-Quilez P, Rojas-González A, Alvarado T, Olivera MJ, et al. Noninvasive diagnosis of vulnerable coronary plaque. World J Cardiol. 2016; 8: 520–533. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5039354/
- Tarkin JM, Dweck MR, Evans NR, Takx RA, Brown AJ, et al. Imaging atherosclerosis. Circ Res. 2016; 118: 750–769. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26892971
- Skinner MP, Yuan C, Mitsumori L, Hayes CE, M.p.s EWR, Nelson JA, et al. Serial magnetic resonance imaging of experimental atherosclerosis detects lesion fine structure, progression and complications in vivo. Nat Med. 1995; 1: 69–73. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7584956
- Toussaint J-F, LaMuraglia GM, Southern JF, Fuster V, Kantor HL. Magnetic Resonance Images Lipid, Fibrous, Calcified, Hemorrhagic, and Thrombotic Components of Human Atherosclerosis in vivo. Circulation. 1996; 94: 932–8.
- Botnar RM, Stuber M, Kissinger KV, Kim WY, Spuentrup E, et al. Noninvasive Coronary Vessel Wall and Plaque Imaging With Magnetic Resonance Imaging. Circulation. 2000; 102: 2582–2587. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11085960
- Fayad ZA, Fuster V. Clinical Imaging of the High-Risk or Vulnerable Atherosclerotic Plaque. Circ Res. 2001; 89: 305–316. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11509446
- Tang TY, Muller KH, Graves MJ, Li ZY, Walsh SR, et al. Iron Oxide Particles for Atheroma Imaging. Arterioscler Thromb Vasc Biol. 2009; 29:1001–1008. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19229073
- Falk E, Shah PK, Fuster V. Coronary Plaque Disruption. Circulation. 1995; 92: 657–671. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7634481
- Virmani R, Kolodgie FD, Burke AP, Finn AV, Gold HK, N T, et al. Atherosclerotic Plaque Progression and Vulnerability to Rupture.
- Hansson GK. Inflammation, Atherosclerosis, and Coronary Artery Disease. N Engl J Med. 2005; 352: 1685–1695. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15843671
- Libby P, DiCarli M, Weissleder R. The Vascular Biology of Atherosclerosis and Imaging Targets. J Nucl Med. 2010; 51(Supplement 1): 335-375. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20395349
- Rudd JHF, Hyafil F, Fayad ZA. Inflammation Imaging in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2009; 29: 1009–1016.
- Reimann C, Brangsch J, Colletini F, Walter T, Hamm B, Botnar RM, et al. Molecular imaging of the extracellular matrix in the context of atherosclerosis. Adv Drug Deliv Rev. 2017; 113:49–60. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27639968
- Virmani R, Burke AP, Farb A, Kolodgie FD. Pathology of the Vulnerable Plaque. J Am Coll Cardiol. 2006; 47(8 Supplement): C13–18. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16631505
- Järveläinen H, Sainio A, Koulu M, Wight TN, Penttinen R. Extracellular Matrix Molecules: Potential Targets in Pharmacotherapy. Pharmacol Rev. 2009; 61:198–223. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2830117/s
- Lutolf MP, Hubbell JA. Synthetic biomaterials as instructive extracellular microenvironments for morphogenesis in tissue engineering. Nat Biotechnol. 2005; 23: 47–55. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15637621
- Grobner T. Gadolinium – a specific trigger for the development of nephrogenic fibrosing dermopathy and nephrogenic systemic fibrosis? Nephrol Dial Transplant. 2006; 21: 1104–1108. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16431890
- George S j., Webb S m., Abraham J l., Cramer S p. Synchrotron X-ray analyses demonstrate phosphate-bound gadolinium in skin in nephrogenic systemic fibrosis. Br J Dermatol. 2010; 163: 1077–1081. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20560953
- Laurent S, Vander Elst L, Henoumont C, Muller RN. How to measure the transmetallation of a gadolinium complex. Contrast Media Mol Imaging. 2010; 5: 305–308. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20803503
- Taupitz M, Stolzenburg N, Ebert M, Schnorr J, Hauptmann R, Kratz H, et al. Gadolinium-containing magnetic resonance contrast media: investigation on the possible transchelation of Gd 3+ to the glycosaminoglycan heparin: GdCM, Glycosaminoglycans and Transchelation. Contrast Media Mol Imaging. 2013; 8:108–116. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23281283
- Narula J, Virmani R, Iskandrian AE. Strategic targeting of atherosclerotic lesions. J Nucl Cardiol. 1999; 6: 81–90. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10070844
- Chatzizisis YS, Coskun AU, Jonas M, Edelman ER, Feldman CL, et al. Role of Endothelial Shear Stress in the Natural History of Coronary Atherosclerosis and Vascular Remodeling: Molecular, Cellular, and Vascular Behavior. J Am Coll Cardiol. 2007; 49: 2379–2393. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17599600
- Gimbrone MA, Topper JN, Nagel T, Anderson KR, Garcia-Cardeña G. Endothelial Dysfunction, Hemodynamic Forces, and Atherogenesisa. Ann N Y Acad Sci. 2000; 902: 230–240. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10865843
- Navab M, Ananthramaiah GM, Reddy ST, Van Lenten BJ, Ansell BJ, et al. The Pathogenesis of Atherosclerosis-The oxidation hypothesis of atherogenesis: the role of oxidized phospholipids and HDL. J Lipid Res. 2004; 45: 993–1007. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15060092
- Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003; 170: 191–203. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14612198
- Winter PM, Morawski AM, Caruthers SD, Fuhrhop RW, Zhang H, et al. Molecular Imaging of Angiogenesis in Early-Stage Atherosclerosis With αvβ3-Integrin–Targeted Nanoparticles. Circulation. 2003; 108: 2270–2274. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14557370
- Galkina E, Ley K. Vascular Adhesion Molecules in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2007; 27: 2292–2301. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17673705
- Kelly KA, Allport JR, Tsourkas A, Shinde-Patil VR, Josephson L, et al. Detection of Vascular Adhesion Molecule-1 Expression Using a Novel Multimodal Nanoparticle. Circ Res. 2005; 96: 327–336. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15653572
- Nahrendorf M, Jaffer FA, Kelly KA, Sosnovik DE, Aikawa E, et al. Noninvasive Vascular Cell Adhesion Molecule-1 Imaging Identifies Inflammatory Activation of Cells in Atherosclerosis. Circulation. 2006; 114: 1504–1511. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17000904
- Briley-Saebo KC, Shaw PX, Mulder WJM, Choi S-H, Vucic E, et al. Targeted Molecular Probes for Imaging Atherosclerotic Lesions With Magnetic Resonance Using Antibodies That Recognize Oxidation-Specific Epitopes. Circulation. 2008; 117: 3206–3215. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18541740
- McAteer MA, Schneider JE, Ali ZA, Warrick 1 Nicholas, Bursill CA, et al. Magnetic Resonance Imaging of Endothelial Adhesion Molecules in Mouse Atherosclerosis Using Dual-Targeted Microparticles of Iron Oxide. Arterioscler Thromb Vasc Biol. 2008 Jan;28(1):77–83. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17962629
- Kwon HM, Sangiorgi G, Ritman EL, McKenna C, Holmes DR, et al. Enhanced coronary vasa vasorum neovascularization in experimental hypercholesterolemia. J Clin Invest. 1998; 101: 1551–1556. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9541483
- Libby P. Inflammation in atherosclerosis. Nature. 2002; 420: 868–874.
- Amirbekian V, Lipinski MJ, Briley-Saebo KC, Amirbekian S, Aguinaldo JGS, et al. Detecting and assessing macrophages in vivo to evaluate atherosclerosis noninvasively using molecular MRI. Proc Natl Acad Sci. 2007; 104: 961–966. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17215360
- Ruehm SG, Corot C, Vogt P, Kolb S, Debatin JF. Magnetic Resonance Imaging of Atherosclerotic Plaque With Ultrasmall Superparamagnetic Particles of Iron Oxide in Hyperlipidemic Rabbits. Circulation. 2001; 103: 415–422. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11157694
- Schmitz SA, Taupitz M, Wagner S, Wolf K-J, Beyersdorff D, et al. Magnetic resonance imaging of atherosclerotic plaques using superparamagnetic iron oxide particles. J Magn Reson Imaging. 2001; 14: 355–361. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11599058
- Kooi ME, Cappendijk VC, Cleutjens KBJM, Kessels AGH, Kitslaar PJEHM, et al. Accumulation of Ultrasmall Superparamagnetic Particles of Iron Oxide in Human Atherosclerotic Plaques Can Be Detected by in vivo Magnetic Resonance Imaging. Circulation. 2003; 107: 2453–2458. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12719280
- Chen J, Tung CH, Mahmood U, Ntziachristos V, Gyurko R, et al. in vivo Imaging of Proteolytic Activity in Atherosclerosis. Circulation. 2002; 105: 2766–2771. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12057992
- Deguchi J, Aikawa M, Tung C-H, Aikawa E, Kim D-E, et al. Inflammation in Atherosclerosis: Visualizing Matrix Metalloproteinase Action in Macrophages in vivo. Circulation. 2006 Jul 4;114(1):55–62. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16801460
- Shipley JM, Wesselschmidt RL, Kobayashi DK, Ley TJ, Shapiro SD. Metalloelastase is required for macrophage-mediated proteolysis and matrix invasion in mice. Proc Natl Acad Sci. 1996; 93: 3942–3946. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC39464/
- Hyafil F, Vucic E, Cornily J-C, Sharma R, Amirbekian V, et al. Monitoring of arterial wall remodelling in atherosclerotic rabbits with a magnetic resonance imaging contrast agent binding to matrix metalloproteinases. Eur Heart J. 2011; 32: 1561–1571. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21118852
- Helske S, Syväranta S, Lindstedt KA, Lappalainen J, Öörni K, et al. Increased Expression of Elastolytic Cathepsins S, K, and V and Their Inhibitor Cystatin C in Stenotic Aortic Valves. Arterioscler Thromb Vasc Biol. 2006; 26: 1791–1798. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16728655
- Majmudar MD, Nahrendorf M. Cardiovascular Molecular Imaging: The Road Ahead. J Nucl Med. 2012; 53: 673–676. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22492729
- Lairez O, Fayad ZA. Imaging of atherosclerosis: Can molecular imaging do more? Arch Cardiovasc Dis. 2013; 106: 551–553.
- Carlier S, Kakadiaris IA, Dib N, Vavuranakis M, O’Malley SM, et al. Vasa vasorum imaging: A new window to the clinical detection of vulnerable atherosclerotic plaques. Curr Atheroscler Rep. 2005; 7: 164–169. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15727733
- Vakonakis I, Campbell ID. Extracellular matrix: from atomic resolution to ultrastructure. Curr Opin Cell Biol. 2007; 19: 578–583. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17942296
- Aumailley M, Gayraud B. Structure and biological activity of the extracellular matrix. J Mol Med. 1998; 76: 253–265. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9535559
- Katsuda S, Kaji T. Atherosclerosis and Extracellular Matrix. J Atheroscler Thromb. 2003; 10: 267–274. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/14718743
- Wight TN, Lara S, Riessen R, Le Baron R, Isner J. Selective deposits of versican in the extracellular matrix of restenotic lesions from human peripheral arteries. Am J Pathol. 1997; 151: 963-973. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9327730
- Wight TN, Merrilees MJ. Proteoglycans in Atherosclerosis and Restenosis: Key Roles for Versican. Circ Res. 2004; 94: 1158–1167. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15142969
- Matter CM, Schuler PK, Alessi P, Meier P, Ricci R, Zhang D, et al. Molecular Imaging of Atherosclerotic Plaques Using a Human Antibody Against the Extra-Domain B of Fibronectin. Circ Res. 2004; 95: 1225–1233.
- Ye F, Jeong E-K, Jia Z, Yang T, Parker D, et al. A Peptide Targeted Contrast Agent Specific to Fibrin-Fibronectin Complexes for Cancer Molecular Imaging with MRI. Bioconjug Chem. 2008; 19: 2300–2303. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19053180
- Mayne R. Collagenous proteins of blood vessels. Arterioscler Thromb Vasc Biol. 1986; 6: 585–593.
- Brodsky B, Persikov AV. Molecular Structure of the Collagen Triple Helix. Adv Protein Chem. 2005; 70: 301–339. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15837519
- Caravan P, Das B, Dumas S, Epstein FH, Helm PA, et al. Collagen-Targeted MRI Contrast Agent for Molecular Imaging of Fibrosis. Angew Chem. 2007; 119: 8319–8321. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17893943
- Helm PA, Caravan P, French BA, Jacques V, Shen L, Xu Y, et al. Postinfarction myocardial scarring in mice: molecular MR imaging with use of a collagen-targeting contrast agent. Radiology. 2008; 247: 788–796. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5410958/
- Jimi S, Sakata N, Matunaga A, Takebayashi S. Low density lipoproteins bind more to type I and III collagens by negative charge-dependent mechanisms than to type IV and V collagens. Atherosclerosis. 1994; 107: 109–116. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7945553
- Khalil MF. Molecular Interactions Leading to Lipoprotein Retention and the Initiation of Atherosclerosis. Arterioscler Thromb Vasc Biol. 2004; 24: 2211–2218. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15472124
- Adiguzel E, Ahmad PJ, Franco C, Bendeck MP. Collagens in the progression and complications of atherosclerosis. Vasc Med. 2009; 14: 73–89. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19144782
- Chen W, Cormode DP, Vengrenyuk Y, Herranz B, Feig JE, et al. Collagen-Specific Peptide Conjugated HDL Nanoparticles as MRI Contrast Agent to Evaluate Compositional Changes in Atherosclerotic Plaque Regression. JACC Cardiovasc Imaging. 2013; 6: 373–384. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/23433925
- Spuentrup E, Ruhl KM, Botnar RM, Wiethoff AJ, Buhl A, et al. Molecular Magnetic Resonance Imaging of Myocardial Perfusion With EP-3600, a Collagen-Specific Contrast Agent: Initial Feasibility Study in a Swine Model. Circulation. 2009; 119: 1768–1775. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/19307474
- Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, et al. Elastin is an essential determinant of arterial morphogenesis. Nature. 1998; 393: 276–280. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9607766
- Karnik SK, Brooke BS, Bayes-Genis A, Sorensen L, Wythe JD, et al. A critical role for elastin signaling in vascular morphogenesis and disease. Development. 2003; 130: 411–423. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12466207
- Kwon GP, Schroeder JL, Amar MJ, Remaley AT, Balaban RS. Contribution of Macromolecular Structure to the Retention of Low-Density Lipoprotein at Arterial Branch Points. Circulation. 2008; 117: 2919–2927. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18506002
- Robert L, Robert AM, Jacotot B. Elastin–elastase–atherosclerosis revisited. Atherosclerosis. 1998; 140: 281–295. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9862271
- Krettek A, Sukhova GK, Libby P. Elastogenesis in Human Arterial Disease: A Role for Macrophages in Disordered Elastin Synthesis. Arterioscler Thromb Vasc Biol. 2003; 23: 582–587. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12615674
- Choudhury Robin P, Valentin F, Badimon Juan J, Fisher Edward A, Fayad Zahi A. MRI and Characterization of Atherosclerotic Plaque. Arterioscler Thromb Vasc Biol. 2002; 22: 1065–1074. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12117718
- Makowski MR, Wiethoff AJ, Blume U, Cuello F, Warley A, et al. Assessment of atherosclerotic plaque burden with an elastin-specific magnetic resonance contrast agent. Nat Med. 2011; 17: 383–388. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21336283
- Fuster V, Stein B, Ambrose JA, Badimon L, Badimon JJ, et al. Atherosclerotic plaque rupture and thrombosis. Evolving concepts. Circulation. 1990; 82(3 Suppl): II47-1159. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2203564
- La Corte ALC, Philippou H, Ariëns RAS. Role of Fibrin Structure in Thrombosis and Vascular Disease. Adv Protein Chem Struct Biol. 2011; 83: 75–127. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/21570666
- Tavora F, Cresswell N, Li L, Ripple M, Burke A. Immunolocalisation of fibrin in coronary atherosclerosis: implications for necrotic core development. Pathology (Phila). 2010; 42: 15‑22. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/20025475
- Botnar RM, Perez AS, Witte S, Wiethoff AJ, Laredo J, et al. in vivo Molecular Imaging of Acute and Subacute Thrombosis Using a Fibrin-Binding Magnetic Resonance Imaging Contrast Agent. Circulation. 2004; 109: 2023–2029. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15066940
- Makowski MR, Forbes SC, Blume U, Warley A, Jansen CHP, et al. in vivo assessment of intraplaque and endothelial fibrin in ApoE−/− mice by molecular MRI. Atherosclerosis. 2012; 222: 43–49.
- Flacke S, Fischer S, Scott MJ, Fuhrhop RJ, Allen JS, et al. Novel MRI Contrast Agent for Molecular Imaging of Fibrin: Implications for Detecting Vulnerable Plaques. Circulation. 2001; 104: 1280–1285. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11551880
- Sirol M, Aguinaldo JGS, Graham PB, Weisskoff R, Lauffer R, et al. Fibrin-targeted contrast agent for improvement of in vivo acute thrombus detection with magnetic resonance imaging. Atherosclerosis. 2005; 182: 79–85. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16115477
- Jackson RL, Busch SJ, Cardin AD. Glycosaminoglucans : molecular properties, protein interactions, and role in physiological processes. Physiol Rev. 1991; 71: 481–539. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2006221
- Salisbury BG, Wagner WD. Isolation and preliminary characterization of proteoglycans dissociatively extracted from human aorta. J Biol Chem. 1981; 256: 8050–8057. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7263639
- Berenson GS, Radhakrishnamurthy B, Srinivasan SR, Vijayagopal P, Dalferes ER. Proteoglycans and Potential Mechanisms Related to Atherosclerosisa. Ann N Y Acad Sci. 1985; 454: 69–78. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3865616
- Taylor KR, Gallo RL. Glycosaminoglycans and their proteoglycans: host-associated molecular patterns for initiation and modulation of inflammation. FASEB J. 2006; 20: 9–22. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16394262
- Lindahl U, Hook M. Glycosaminoglycans and their binding to biological macromolecules. Annu Rev Biochem. 1978; 47: 385–417. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/354500
- Turnbull J, Powell A, Guimond S. Heparan sulfate: decoding a dynamic multifunctional cell regulator. Trends Cell Biol. 2001; 11: 75–82. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11166215
- Nugent MA. Heparin sequencing brings structure to the function of complex oligosaccharides. Proc Natl Acad Sci. 2000; 97: 10301–10303. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10984527
- Evanko SP, Raines EW, Ross R, Gold LI, Wight TN. Proteoglycan distribution in lesions of atherosclerosis depends on lesion severity, structural characteristics, and the proximity of platelet-derived growth factor and transforming growth factor-beta. Am J Pathol. 1998; 152: 533–546. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1857967/
- Stevens RL, Colombo M, Gonzales JJ, Hollander W, Schmid K. The glycosaminoglycans of the human artery and their changes in atherosclerosis. J Clin Invest. 1976; 58: 470–481. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC333202/
- Ballinger ML, Nigro J, Frontanilla KV, Dart AM, Little PJ. Regulation of glycosaminoglycan structure and atherogenesis. Cell Mol Life Sci CMLS. 2004; 61: 1296–1306. pubMed: https://www.ncbi.nlm.nih.gov/pubmed/15170508
- Kolodgie FD, Burke AP, Farb A, Weber DK, Kutys R, et al. Differential Accumulation of Proteoglycans and Hyaluronan in Culprit Lesions: Insights Into Plaque Erosion. Arterioscler Thromb Vasc Biol. 2002; 22: 1642–1648. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12377743
- O'Brien KD, Olin KL, Alpers CE, Chiu W, Ferguson M, et al. Comparison of Apolipoprotein and Proteoglycan Deposits in Human Coronary Atherosclerotic Plaques: Colocalization of Biglycan With Apolipoproteins. Circulation. 1998; 98: 519–527. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9714108
- Merrilees MJ, Beaumont B, Scott LJ. Comparison of deposits of versican, biglycan and decorin in saphenous vein and internal thoracic, radial and coronary arteries: correlation to patency. Coron Artery Dis. 2001; 12:7–16. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11211169
- Radhakrishnamurthy B, Srinivasan SR, Vijayagopal P, Berenson GS. Arterial wall proteoglycans‑biological properties related to pathogenesis of atherosclerosis. Eur Heart J. 1990; 11: 148‑157. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2226523
- Reitsma S, Slaaf DW, Vink H, van Zandvoort MAMJ, oude Egbrink MGA. The endothelial glycocalyx: composition, functions, and visualization. Pflüg Arch - Eur J Physiol. 2007; 454: 345–59. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17256154
- Tarbell JM, Cancel LM. The glycocalyx and its significance in human medicine. J Intern Med. 2016; 280:97–113. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/26749537
- Selleck SB. Proteoglycans and pattern formation: sugar biochemistry meets developmental genetics. Trends Genet. 2000; 16: 206–212. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10782114
- Hileman RE, Fromm JR, Weiler JM, Linhardt RJ. Glycosaminoglycan-protein interactions: definition of consensus sites in glycosaminoglycan binding proteins. Bioessays. 1998; 20: 156–167. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9631661
- Wight TN. The extracellular matrix and atherosclerosis. Curr Opin Lipidol. 1995; 6: 326.
- Scott JE. Structure and function in extracellular matrices depend on interactions between anionic glycosaminoglycans. Elsevier; 2001; 49: 284-289. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11428163
- Srinivasan SR, Xu J-H, Vijayagopal P, Radhakrishnamurthy B, Berenson GS. Low-density lipoprotein binding affinity of arterial chondroitin sulfate proteoglycan modulates cholesteryl ester accumulation in macrophages. Atherosclerosis. 1994; 109: 97.
- Hurt-Camejo E, Olsson U, Wiklund O, Bondjers G, Camejo G. Cellular Consequences of the Association of ApoB Lipoproteins With Proteoglycans. Arterioscler Thromb Vasc Biol. 1997; 17: 1011–1017. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9194748
- Camejo G, Olofsson SO, Lopez F, Carlsson P, Bondjers G. Identification of Apo B-100 segments mediating the interaction of low density lipoproteins with arterial proteoglycans. Arterioscler Thromb Vasc Biol. 1988; 8: 368–377. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3395272
- Skålén K, Gustafsson M, Rydberg EK, Hultén LM, Wiklund O, et al. Subendothelial retention of atherogenic lipoproteins in early atherosclerosis. Nature. 2002; 417: 750–754. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12066187
- Borén J, Olin K, Lee I, Chait A, Wight TN, et al. Identification of the principal proteoglycan-binding site in LDL. A single-point mutation in apo-B100 severely affects proteoglycan interaction without affecting LDL receptor binding. J Clin Invest. 1998; 101: 2658–2664. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9637699
- Imberty A, Lortat-Jacob H, Pérez S. Structural view of glycosaminoglycan–protein interactions. Carbohydr Res. 2007; 342: 430–439. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17229412
- Busch C, Dawes J, Pepper DS, Wasteson A. Binding of platelet factor 4 to cultured human umbilical vein endothelial cells. Thromb Res. 1980; 19: 129–137. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7444850
- Stuckey JA, Charles RSt, Edwards BFP. A model of the platelet factor 4 complex with heparin. Proteins Struct Funct Bioinforma. 1992 ; 14: 277–287.
- Perollet C, Han ZC, Savona C, Caen JP, Bikfalvi A. Platelet Factor 4 Modulates Fibroblast Growth Factor 2 (FGF-2) Activity and Inhibits FGF-2 Dimerization. Blood. 1998; 91: 3289–3299. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9558385
- Petersen F, Brandt E, Lindahl U, Spillmann D. Characterization of a Neutrophil Cell Surface Glycosaminoglycan That Mediates Binding of Platelet Factor 4. J Biol Chem. 1999; 274: 12376–12382. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10212210
- Jin L, Abrahams JP, Skinner R, Petitou M, Pike RN, Carrell RW. The anticoagulant activation of antithrombin by heparin. Proc Natl Acad Sci. 1997; 94: 14683–14688. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9405673
- Li W, Johnson DJD, Esmon CT, Huntington JA. Structure of the antithrombin–thrombin–heparin ternary complex reveals the antithrombotic mechanism of heparin. Nat Struct Mol Biol. 2004; 11: 857–862. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15311269
- Raman R, Venkataraman G, Ernst S, Sasisekharan V, Sasisekharan R. Structural specificity of heparin binding in the fibroblast growth factor family of proteins. Proc Natl Acad Sci. 2003; 100:2357–2362. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12604799
- Guerrini M, Agulles T, Bisio A, Hricovini M, Lay L, et al. Minimal Heparin/Heparan Sulfate Sequences for Binding to Fibroblast Growth Factor-1. Biochem Biophys Res Commun. 2002; 292: 222–230. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11890696
- Capila I, Linhardt RJ. Heparin–Protein Interactions. Angew Chem Int Ed. 2002; 41: 390–412. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12491369
- Desai UR, Petitou M, Björk I, Olson ST. Mechanism of Heparin Activation of Antithrombin Role of Individual Residues of the Pentasaccharide Activating Sequence in the Recognition of Native and Activated States of Antithrombin. J Biol Chem. 1998; 273: 7478–87. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9516447
- Maimone MM, Tollefsen DM. Structure of a dermatan sulfate hexasaccharide that binds to heparin cofactor II with high affinity. J Biol Chem. 1990; 265: 18263–18271. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/2211700
- Liaw PCY, Becker DL, Stafford AR, Fredenburgh JC, Weitz JI. Molecular Basis for the Susceptibility of Fibrin-bound Thrombin to Inactivation by Heparin Cofactor II in the Presence of Dermatan Sulfate but Not Heparin. J Biol Chem. 2001; 276: 20959–20965. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/11294849
- Uca YO, Hallmann D, Hesse B, Seim C, Stolzenburg N, Pietsch H, et al. Microdistribution of Magnetic Resonance Imaging Contrast Agents in Atherosclerotic Plaques Determined by LA-ICP-MS and SR‑µXRF Imaging. Invest Radiol. 2020 Mar 24 (Manuscript submitted for publication)
- Collingwood JF, Adams F. Chemical imaging analysis of the brain with X-ray methods. Spectrochim Acta Part B At Spectrosc. 2017; 130: 101–18.
- Hare D, Austin C, Doble P. Quantification strategies for elemental imaging of biological samples using laser ablation-inductively coupled plasma- mass spectrometry. Analyst. 2012; 137: 1527–1537. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/22314636
- Kubaski F, Osago H, Mason RW, Yamaguchi S, Kobayashi H, et al. Glycosaminoglycans detection methods: Applications of mass spectrometry. Mol Genet Metab. 2017; 120: 67–77. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/27746032
- Caravan P. Strategies for increasing the sensitivity of gadolinium based MRI contrast agents. Chem Soc Rev. 2006; 35: 512-523. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16729145
- Ziegler A, Seelig J. Binding and Clustering of Glycosaminoglycans: A Common Property of Mono- and Multivalent Cell-Penetrating Compounds. Biophys J. 2008; 94: 2142–2149. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18065465
- Parish CR. The role of heparan sulphate in inflammation. Nat Rev Immunol. 2006; 6: 633–643. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16917509
- Li Q, Park PW, Wilson CL, Parks WC. Matrilysin Shedding of Syndecan-1 Regulates Chemokine Mobilization and Transepithelial Efflux of Neutrophils in Acute Lung Injury. Cell. 2002; 111: 635–646. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/12464176
- Bernfield M, Götte M, Park PW, Reizes O, Fitzgerald ML, et al. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999; 68: 729–777. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/10872465
- Handel TM, Johnson Z, Crown SE, Lau EK, Proudfoot AE. Regulation of Protein Function by Glycosaminoglycans—as Exemplified by Chemokines. Annu Rev Biochem. 2005; 74: 385–410. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15952892
- Fischer JW, Steitz SA, Johnson PY, Burke A, Kolodgie F, et al. Decorin Promotes Aortic Smooth Muscle Cell Calcification and Colocalizes to Calcified Regions in Human Atherosclerotic Lesions. Arterioscler Thromb Vasc Biol. 2004; 24: 2391–2396. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15472131
- McDonald JA. Extracellular matrix assembly. Annu Rev Cell Biol. 1988;4(1):183–207.
- Costa DS da, Reis RL, Pashkuleva I. Sulfation of Glycosaminoglycans and Its Implications in Human Health and Disorders. Annu Rev Biomed Eng. 2017; 19: 1–26. PubMed: https://www.ncbi.nlm.Fnih.gov/pubmed/28226217
- Thacker BE, Xu D, Lawrence R, Esko JD. Heparan sulfate 3-O-sulfation: A rare modification in search of a function. Matrix Biol. 2014; 35: 60–72. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24361527
- Busby TF, Argraves WS, Brew SA, Pechik I, Gilliland GL, et al. Heparin Binding by Fibronectin Module III-13 Involves Six Discontinuous Basic Residues Brought Together to Form a Cationic Cradle. J Biol Chem. 1995; 270: 18558–18562. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7629186