
More Information
Submitted: November 10, 2020 | Approved: January 14, 2021 | Published: January 15, 2021
How to cite this article: Smith GA. New insights from cardiac muscle applied to skeletal muscle. J Cardiol Cardiovasc Med. 2021; 6: 007-013.
DOI: 10.29328/journal.jccm.1001109
Copyright License: © 2021 Smith GA. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Abbreviations: MyBP-C: Myosin Binding Protein-C (was called protein-C); TNN: Gene Name in Database; TnC: Troponin-C; TnI: Troponin-I; Tm: Tropomyosin; TnT: Troponin T; pCa50: The point with 50% activation

New insights from cardiac muscle applied to skeletal muscle
Gerry A Smith*
Retired, University of Cambridge, Cambridge CB2 9NJ, UK
*Address for Correspondence: Gerry A Smith, University of Cambridge, Retired, 14, Lantree Crescent, Trumpington, Cambridge CB2 9NJ, UK, Tel: 01223 515394; Email: [email protected]
I have recently described the origin of the second Ca2+ binding in the triggering of contractile activity in cardiac myofibrils that is the origin of the Ca2+ Hill coefficient of 2 for the ATPase. This site is not a simple protein binding site and cannot be measured by 45Ca2+ binding. The myofibril protein unit requirements are described by me and so are the consequences of disruption of the function of these units and the related medical outcomes. The purpose of this paper is to review the topic and extend the reasoning to the function of skeletal muscle and cite the literature that supports this.
In muscle activation Ca2+ binds to the activating subunit troponin-C (TnC) which changes its interaction with another subunit troponin-I (TnI) which changes the interaction of this with yet another subunit tropomyosin (Tm). The consequence of this complex activity is the unblocking of interaction of the myosin light chain (LC), ATP bound, with the actin chain allowing the formation of a cross-bridge between the thick and thin filaments and the resulting ATPase activity giving muscle contraction and/or force. The cycle is completed by replacement of the ATPase products (ADP and phosphate Pi) by the binding of MgATP to the myosin light chain. This is the story accepted to date in all striated muscle systems. However in the cardiac system I first suggested in 2001 [1] that this was not all embracing and mooted that a further Ca2+ binding was required to satisfy the observed cooperativity (raised Hill coefficient for Ca2+ activation > 1). This I have recently revisited [2] to show the implication of yet another myofibril subunit in the Ca2+ activation of myocyte contraction, i.e. the myosin binding protein-C (MyBP-C) [2-4] and applied this to the cardiac system [5], and thus demonstrating the origin of many cardiomyopathies, particularly hypertropy [5]. The key to these results is that the MgATP bound myosin is the fully relaxed state, it is not used in the functioning of the cross-bridge and in the unperturbed system the presence of intact cMyBP-C ensures this. There are many references to the cMyBP-C acting to modulate the Ca2+ sensitivity of the cardiac muscle but absolutely none give clear mechanistic explanation of how this occurs. My recent papers show that along with the cTnI the cMyBP-C ensure the Myosin bound MgATP has to become myosin bound CaATP before the cross-bridge can form. In the heart this binding of Ca2+ requires a concentration [Ca2+] well above that required by the TnC binding and is also inhibited competitively by Mg2+. Any failure of this concerted action of the subunits, cTnI and cMyBP-C, results in the binding of Ca2+ by cTnC becoming the sole requirement for activation of contraction, this produces some contraction at much lower [Ca2+] and hence incomplete relaxation in diastole. The result of incomplete relaxation in diastole is chronic tension that is transmitted to another myofibril component, the giant protein titin, this tension acts at the spring region of the titin releasing a myocyte growth factor the result of which is unwanted growth, i.e. hypertrophy.
For considering extending the above findings to skeletal myofibrils the key lead in citations involve those that deplete the sarcomere of subunits by the use of EDTA to remove all bound divalent cations from their structural binding sites. This also includes the substitution with differently resourced units or retention of specific proteins [6,7]. I initially lay the ground by giving a comprehensive review of the three subunits intimately involved.
In this study I concentrate on three proteins that are essential components of the Ca2+ stimulated control of muscle contraction. The proteins are the Ca2+ binding activating unit Troponin-C (TnC), the inbibitory unit Troponin-I (TnI) and myosin binding protein-C (MyBP-C), table 1. These all show isoforms specific to adult muscle type, fast and slow skeletal and cardiac. The gene name and number for TnI and MyBP-C are in agreement, three genetic versions slow skeletal, fast skeletal and cardiac but only two, fast skeletal and slow/cardiac for TnC.
| Table 1:The isoforms of the key sarcomere subunits (from Uniprot). | |||
| Protein isoforms | Slow skeletal muscle ssTnI |
Fast Skeletal muscle fsTnI |
Cardiac muscle cTnI |
| Gene | TNNI1 | TNNI2 | TNNI3 |
| Chromosome | 1q31.3 | 11p15.5 | 19q13.4 |
| Protein isoforms | Slow Skeletal Muscle ssMyBP-C |
Fast Skeletal Muscle fsMyBP-C |
Cardiac muscle cMyBP-C |
| Gene | MYBPC1 | MYBPC2 | MyBPC3 |
| Chromosome | 12q23.2 | 19q13.33 | 11p11.2 |
| Protein isoforms | Slow Skeletal Muscle ssTnC |
Fast Skeletal Muscle fsTnC |
Cardiac muscle cTnC |
| Gene | TNNC1 | TNNC2 | TNNC1 |
| Chromosome | 3p21.1 | 20q13.12 | 3p21.1 |
| Note, the ssTnC and cTnC are the same protein. | |||
The notable differences between these protein isoforms
TnI comparisons: The most obvious difference are between the cardiac and the other (skeletal) versions. The cardiac has an extended N terminal sequence that folds and binds to the inhibitory region (residues 137-148) Figure 1.
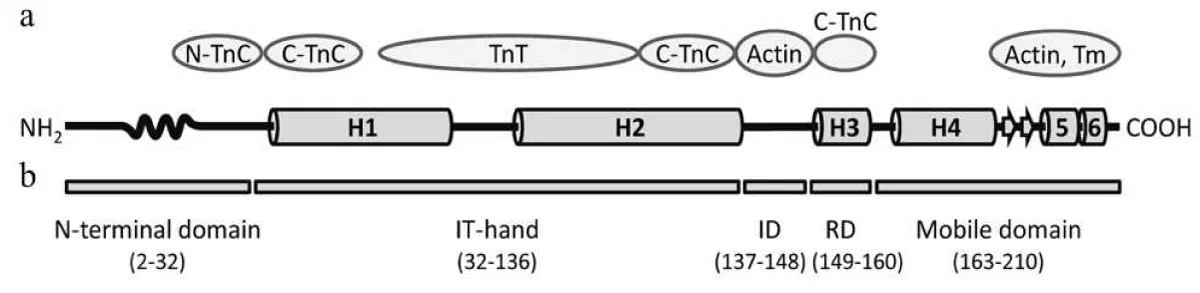
Figure 1: Domain structure of human cTnI [8]. a) Scheme of the cTnI secondary structure. The N-terminal domain of TnI is residues 2-32, the α-helices H1-H4 in the TnI, and the mobile domain H4-H6. The wavy curve represents the Xaa-Pro region of cTnI (residues 12-18) forming a proline helix. Short β-strands 1and 2 are marked by arrows. Proteins of the thin filament that interact with the relevant regions of the cTnI molecule are indicated as ovals. N-TnC and C-TnC, N- and C-terminal domains of TnC, respectively; Tm, tropomyosin. b) Domain organization of cTnI. ID, inhibitory domain; RD, regulatory domain.
Layland, et al. [9] report “binding of Ca2+ to cTnC during systole induces conformational changes that relieve the inhibitory influence of cardiac cTnI”. More correctly put this should read “binding of Ca2+ to troponin C and myosin ATP….”
Phosphorylation of cTnI alters its function, in particular the threonine of the cardiac version (residue 142 or 3 or 4 depending on the reference used) which undergoes phosphorylation by PKC-βII under diastolic stretch, the Frank-Starling law [3,4,10], all the TnIs also get serine phosphorylated in various places altering function.
Mutations of TnI
To date, no human disease has been reported with mutations in ssTnI [11]. However mutations in the fsTnI gene have been found to cause myopathy and distal arthrogryposis (DA). A missense mutation R174Q, a nonsense mutation (premature stop codon R156X), and three in-frame deletion mutations ΔE167, ΔK175 and ΔK176 have been reported in DA patients [12]. The mutations associated with DA are all in the C-terminal actin-tropomyosin binding domain. Many mutations of cTnI have been found to cause hyper, restrictive, and dilated myopathies [5,13,14].
MyBP-C comparisons
The name, now changed from protein-C to myosin binding protein-C arises from the first findings that it binds to the S2 region of myosin, Figure 2. The last realised unit of the sarcomere can only now be recognised as important as TnC in triggering of contraction, if not more so. It is unfortunate that the binding to actin has been so overlooked in many studies. The FHL-1 gene binding couple with the titin binding is probably reflected in myofibril growth, hypertrophy, as is the FHOD-3 binding.
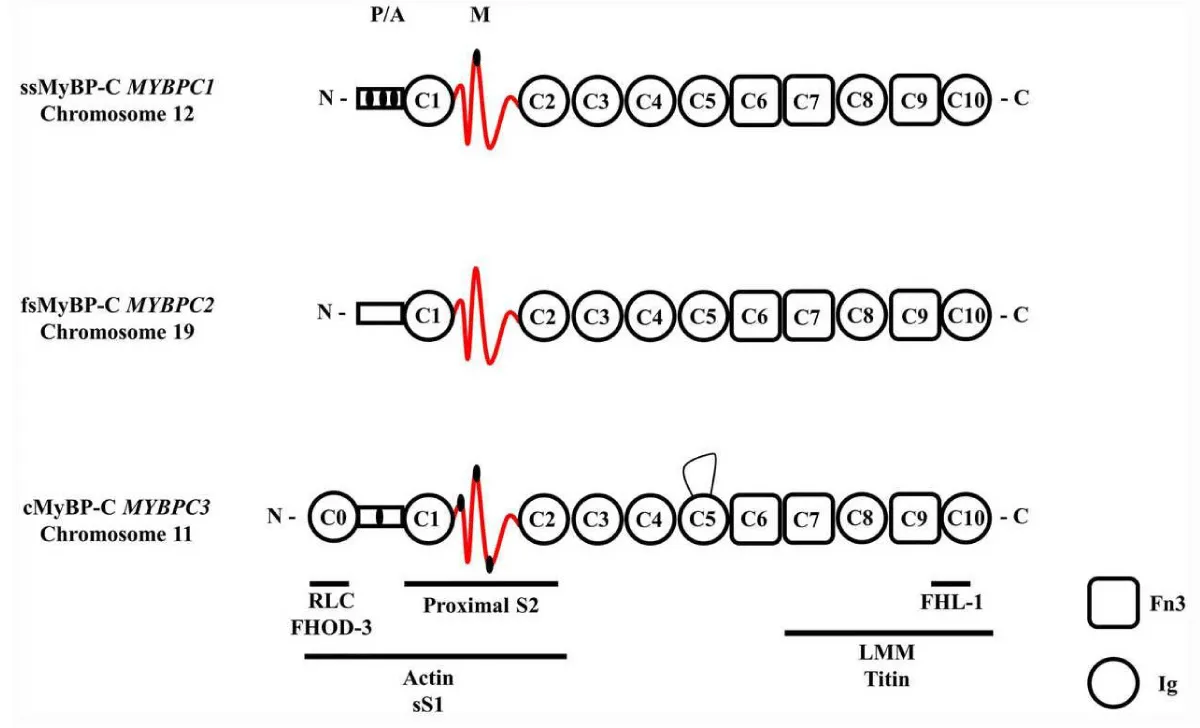
Figure 2: Schematic diagram of full length slow skeletal (ss), fast skeletal (fs) and cardiac (c) MyBPC paralogs. Each isoform comprises three Fn3 domains and seven or eight Ig domains. The known binding partners and positions are indicated by the horizontal stripes below. Note the regulatory phosphorylation sites in the P/A and M domain of the ssMyBP-C and cMyBP-C paralogs are indicated by small black ellipses. The cMyBP-C has an additional 28 amino acid loop in the C5 domain (15,) and one clearly obviously wrong reference [17].
TnC comparisons
A comprehensive review by Katrukha [8] has many leading references within (Figure 3). All TnC forms have four Ca2+-binding EF-hands that are combined pairwise into the N-terminal (sites I and II) and C-terminal (sites III and IV) globular domains. In the fast skeletal isoform of TnC, all four EF-hands are able to bind Ca2+ or Mg2+. Only the N-terminal domain EF-hands I and II have a lower affinity but high selectivity for Ca2+ and both play a crucial role in the regulation of muscle contraction. Sites III and VI are high affinity, are ion bound under all physiological conditions and structural and bind TnC to the sarcomere.
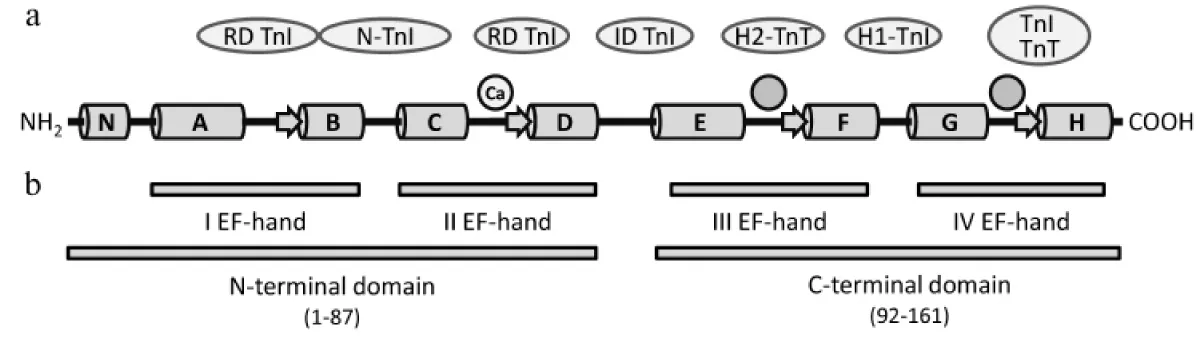
Figure 3: Domain structure of human cardiac/slow skeletal cTnC. The α-Helices N and A-H are represented by cylinders, β-strands by arrows. Two circles without designation mark the structural high affinity sites III and IV. The circle designated with “Ca” represents the one high-specificity region that binds Ca2+, Site II. Proteins of the thin filament that interact with the relevant regions of the troponin-T (TnT) molecule are indicated in ovals. N-TnI, N-terminal domain of TnI; RD, regulatory domain; ID, inhibitory domain.
A few amino acid substitutions in the first EF-hand of the c/ssTnC isoform (site I) impede the ion binding so that c/ssTnC has only three Ca2+-binding sites, only one active in contraction stimulation.
The activation binding of Ca2+ to the two types of TnC is well defined by Lee [17] and has been resolved for some time [18-20]. In the heart the single activating Ca2+ binding site in cTnC gives rise to a Ca2+ Hill coefficient of 2 when measured carefully on the completely intact system. This cooperativity of a second site with the single TnC site II is explained as arising from an exchange of Mg2+ for Ca2+ on the myosin light chain controlled by cMyBP-C [1-4]. For the fast skeletal muscle the situation is more muddled as the Hill coefficient, especially when measuring tension can be much higher than the value of 3 that is anticipated if the two fsTnC sites work with the fsMyBP-C in the same way as cardiac [21].
The sarcolema complex
Scheme 1, the cross-bridge cycle
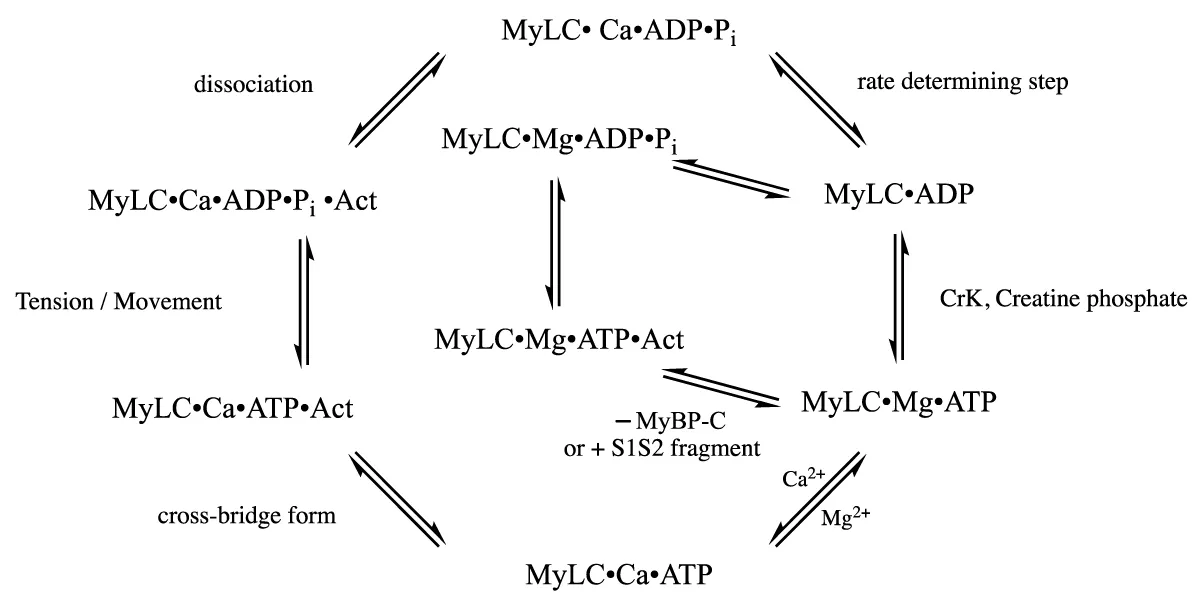
Scheme 1: The cross-bridge cycle.
The Actin filament carries the troponin complex, TnC troponin-C (the Ca2+ binding activation), TnI (the inhibitory unit)whose inhibitory function is relieved on Ca2+ activation and Tm (the tropomyosin) which is moved away to allow the MyLC-Actin cross-bridge to form and last, the TnT troponin T.
The ATPase protein complex consists of MyBP-C which through its N-terminus (S1S2) binds both thick filament (Myosin) and thin filament (Actin). Creatine Kinase is bound to the MyBP-C. The MyBP-C S1S2 fragment when added binds to the actin and myosin, scheme 1, in the same way as the whole in the spaces between the MyBP-C units. The fragment does not have the MgATP inhibitory function of the whole and bypasses the Ca2+ swap for Mg2+ as does the MyBP-C removal, this cation swap obeys normal competitive binding. The filaments are associated along length with the giant protein Titin which is structural and reacts to chronic stress with the release of sarcolemal growth factors.
In the light of my previous studies on the cardiac system [1-5], in particular the effect of removal of cTnC, Figure 4 [7], my start point was the same treatment given earlier to skeletal muscle by Brandt, et al. [6]. The data quoted are nearly all presented as fractional tension v pCa charts the shifts and slope changes of which are informative of what is happening.
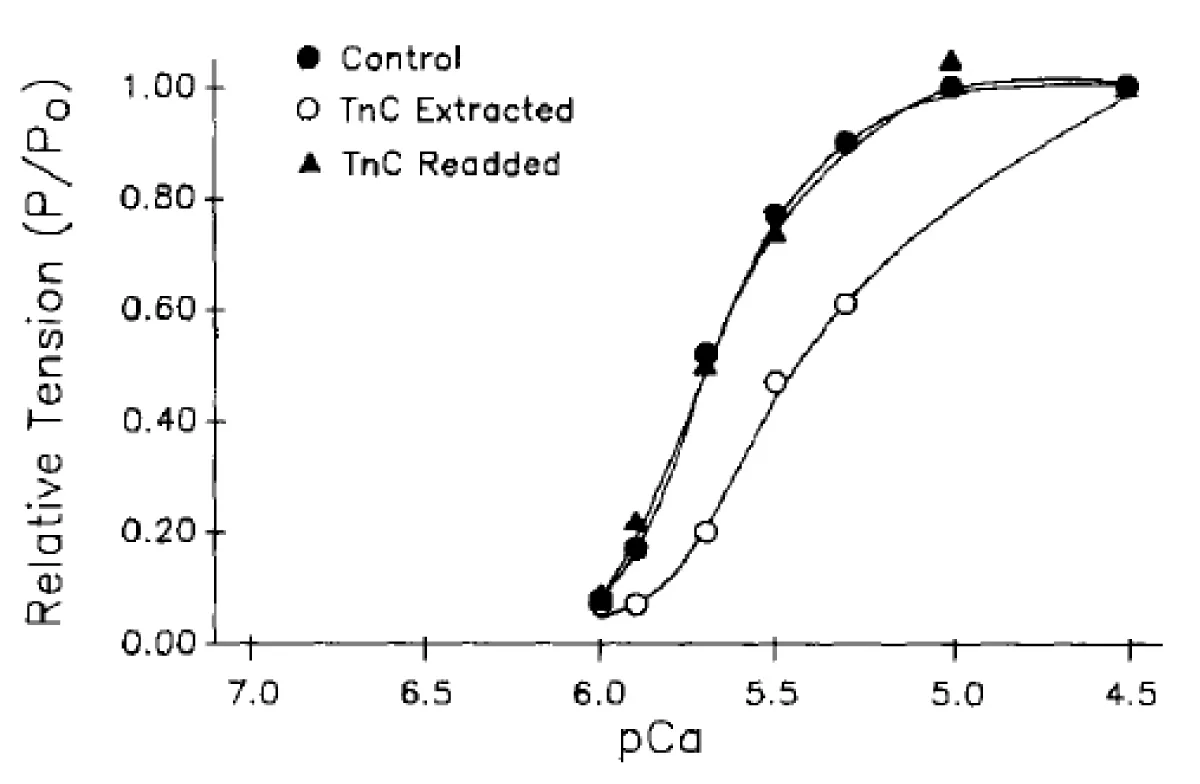
Figure 4: The effects of cTnC extraction on cardiac myofibrils [7]. Data from a myocyte before cTnC extraction (●), after partial extraction of cTnC (○), and finally, with readdition of cTnC (▲). After extraction, tension at pCa 4.5 was 0.49 of control Po; with readdition, maximum tension returned to 0.75 of control Po.
The extraction of cTnC
First I quote from Hofmann the results obtained with the cardiac system, Figure 4 (and later Figures 8 and 9 for comparison with their other data), I follow with the addition of an N-terminal fragment of MyBP-C to cardiac myofibrils, Figure 5, to introduce the MyBP-C extraction problem with other data and The Brandt data giving figure 6.
The addition of the N-terminal fragment of MyBP-C, C1mC2, to cardiac myofilaments
The addition of the N-terminal fragments of MyBP-C, Figure 5 [22] to the cardiac myofibrils changes the response of the system activation to the level of [Ca2+] defining the importance of MyBP-C in the control mechanism.
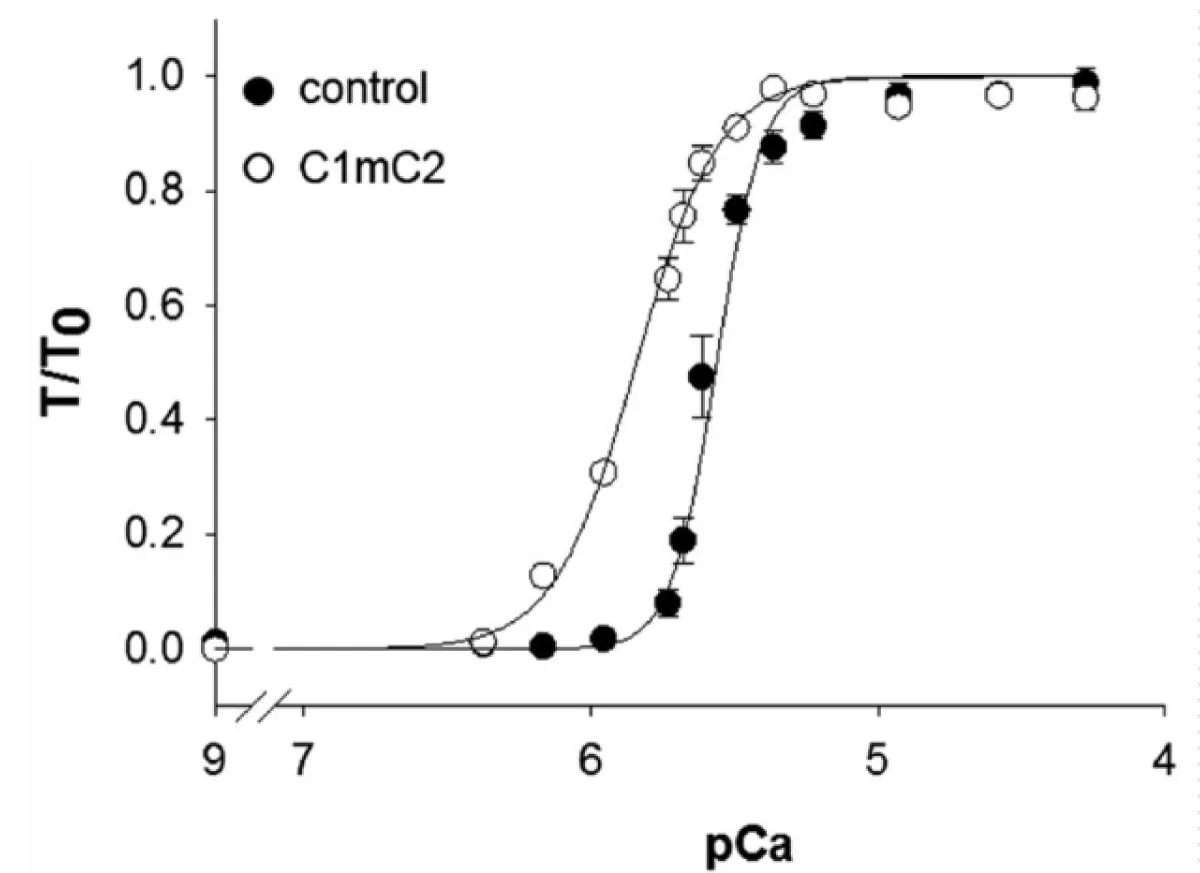
Figure 5: Calcium dependence of force in the absence (●) and the presence (○) of 2μmol/L N-terminal fragment C1mC2 [22].
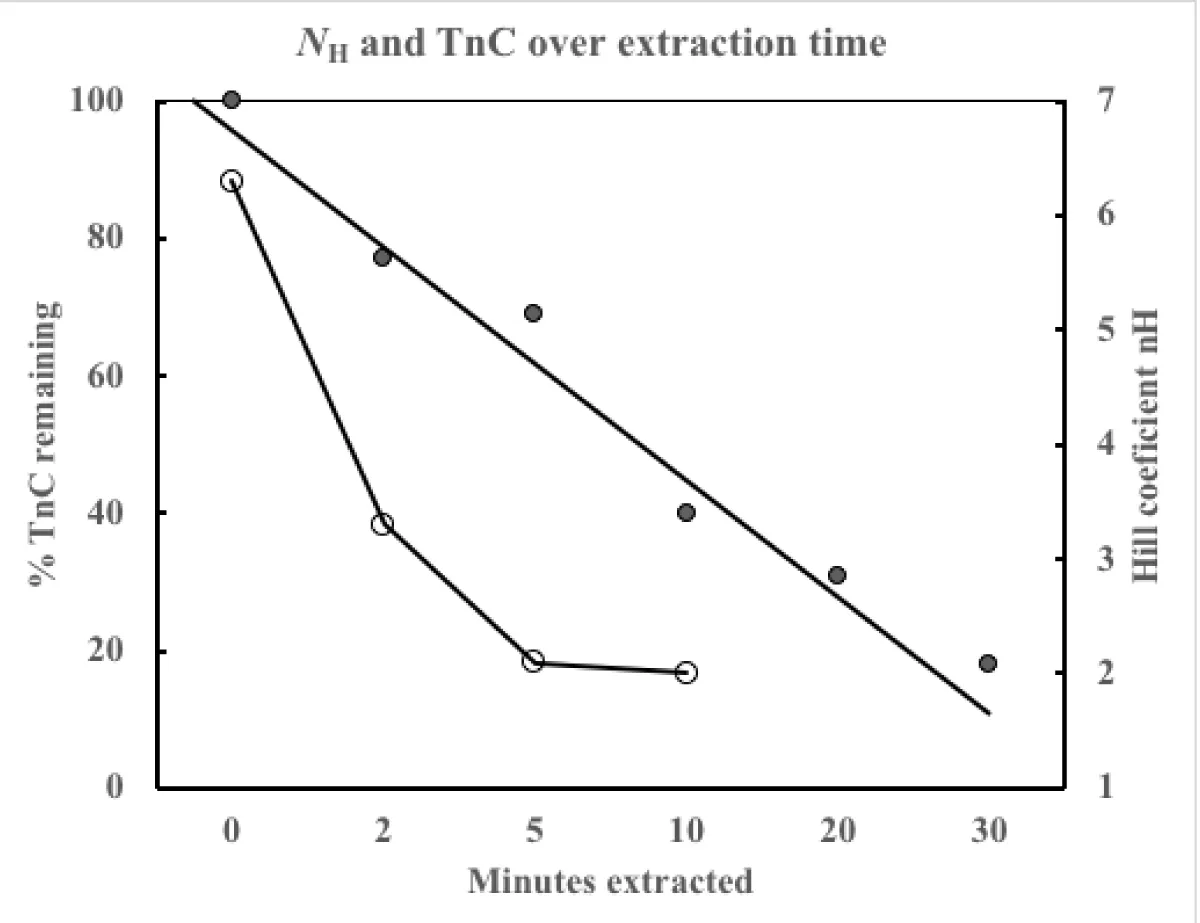
Figure 6: The filled circles (●) are the % TnC remaining, the open circles (○) the calculated Hill coefficient nH which asymptotes to a value of 2.
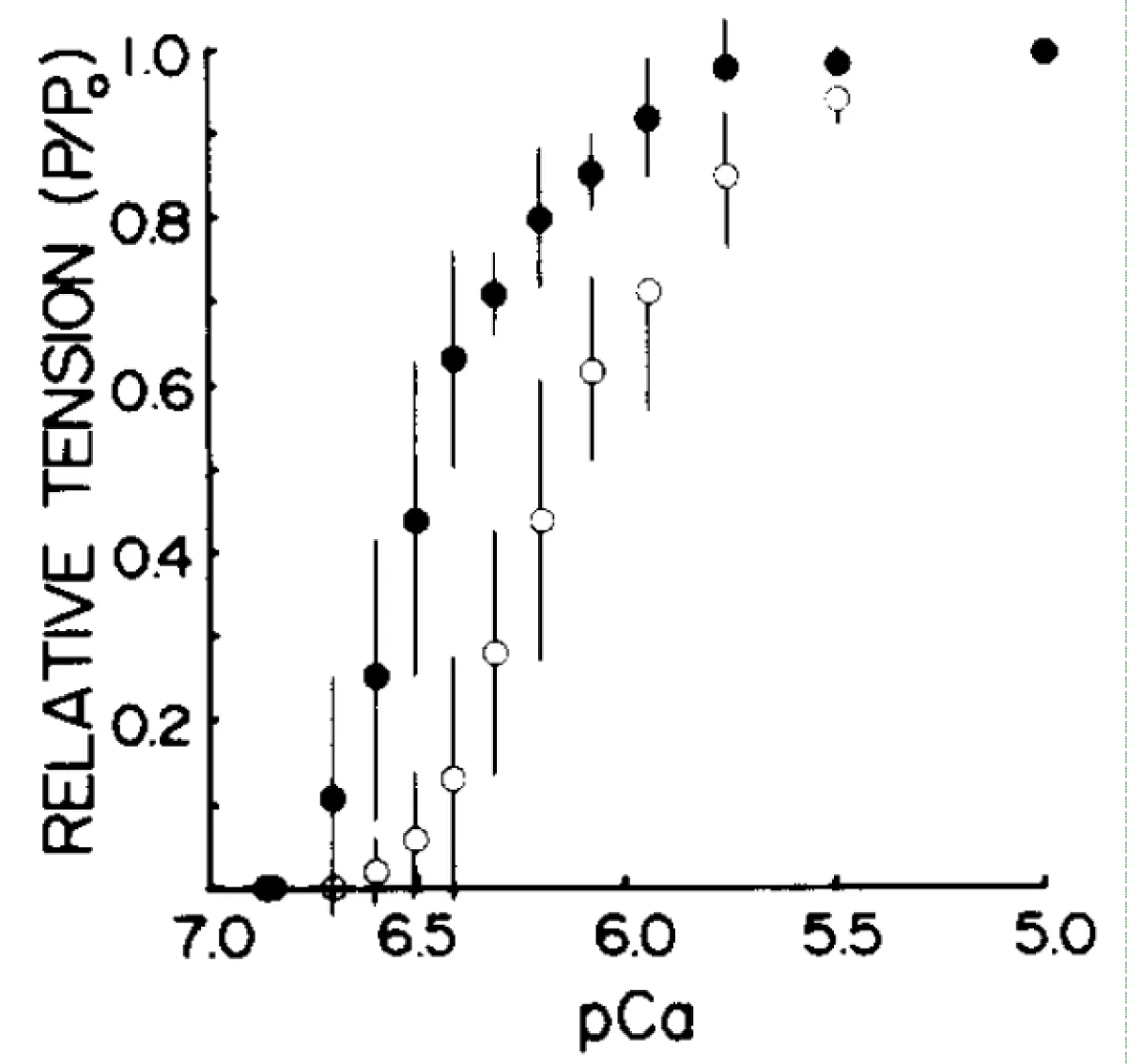
Figure 7: Plots of the dependence of tension on pCa before (●) and after (○) partial extraction of TnC tension at each pCa is expressed as a fraction of the tension developed by the same fiber at pCa 5.0. Error bars indicate SD.
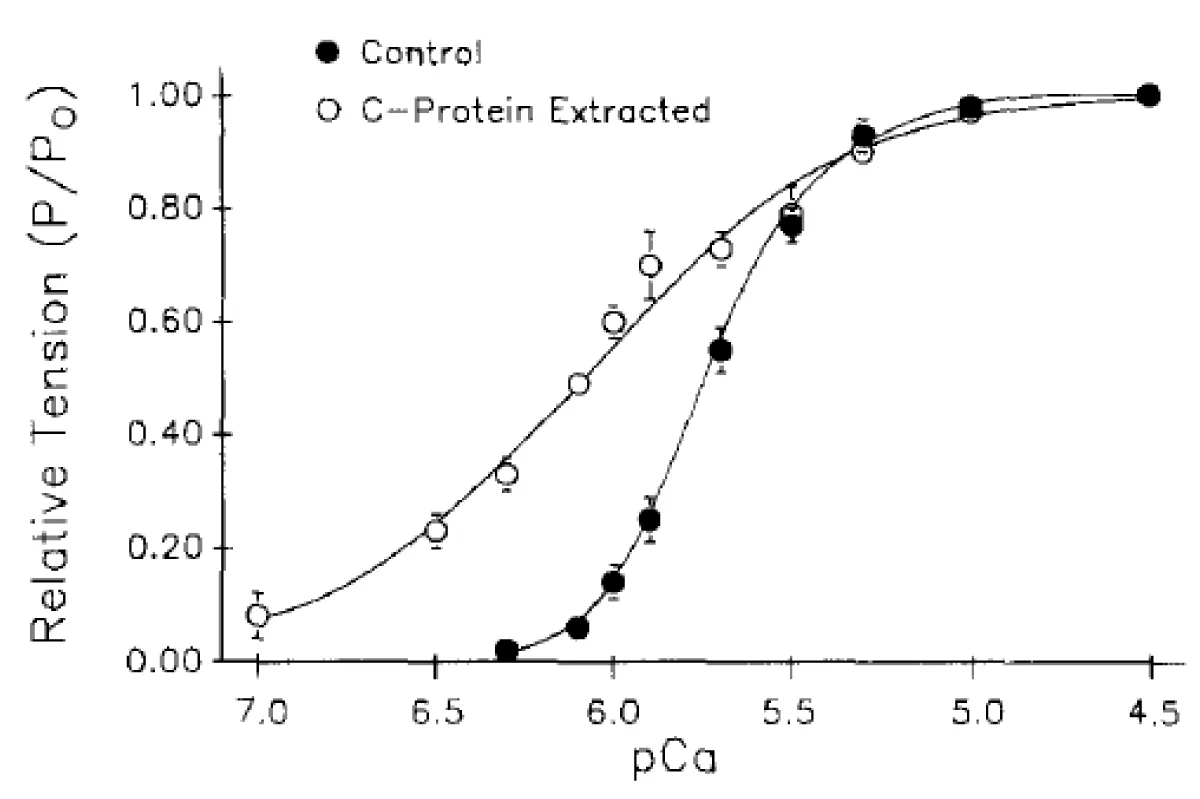
Figure 8: Tension-pCa data obtained before (●) and after (○) partial extraction of cMyBP-C from rat ventricular myocytes (with added cTnC). Symbols and error bars indicate means ± SEM from 17 myocytes. The data are plotted as relative tension-pCa relationships.
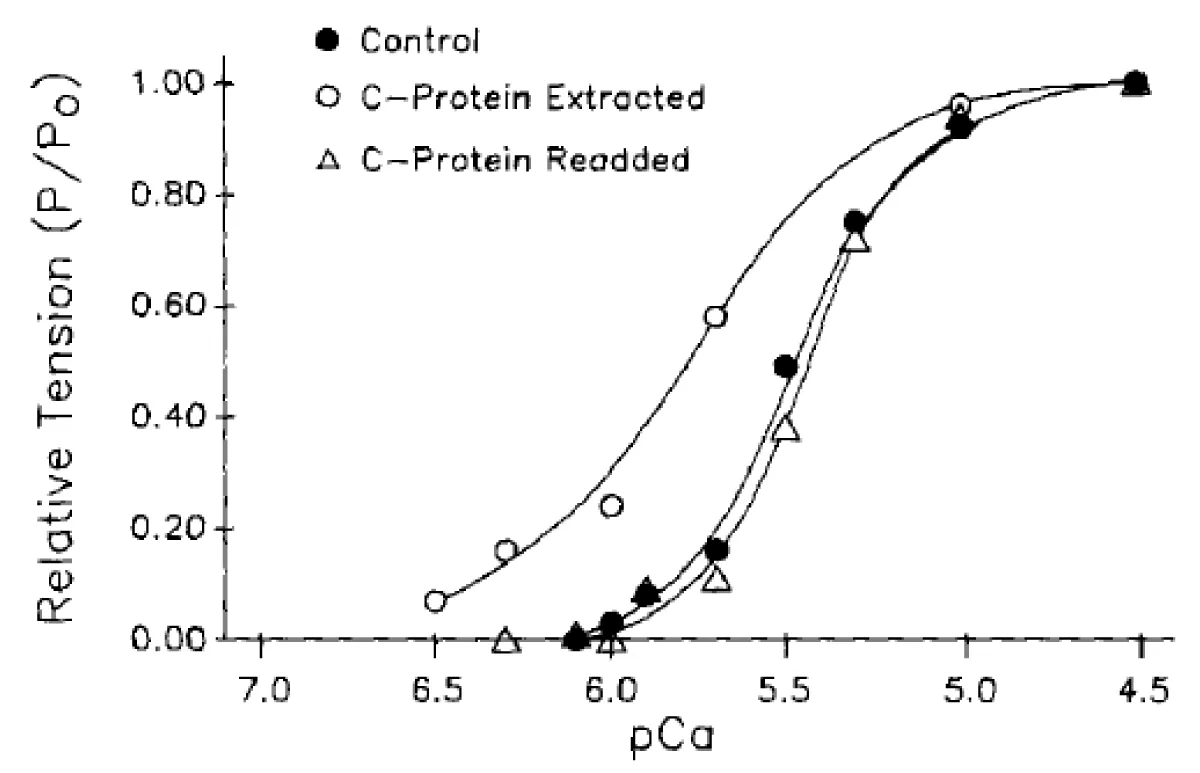
Figure 9: Reversibility of cMyBP-C extraction as above.
I now realise how the addition of N-terminal fragments give this result. It was initially believed that the MyBP-C binding to the myosin S2 region [23] was being displaced by the fragment, this is not true as the Km for the Ca2+ independent activation is much higher than the concentration used. In fact the MyBP-C binds to actin as well as myosin [24] and is located along the filaments spaced out with many more binding sites on both filaments unoccupied. The N-terminal fragment, especially with cardiac filaments, binds both the actin and the myosin [25], with the many unoccupied binding sites the fragment binds to these and as it lacks the MgATP blocking of the whole subunit it allows the ATPase to function without the Mg2+-Ca2+ exchange. The curve is shifted to lower [Ca2+], towards the Km for TnC, and the cooperativity is lost, the Hill coefficient goes to 1.
The extraction of fsTnC from skeletal myofibrils
In this early study the ion binding to the structural sites of TnC is removed by the indiscriminate binding of Mg2+ and Ca2+ by the low selectivity chelator EDTA [6]. This allows the dissociation of the unit from the sarcomere and its washing away. Doing stopped assays over a time course gave the data in figure 6 and table 2. Note the tension developed greatly reduces with extraction time limiting the observations possible to the earlier times.
| Table 2 | ||||||
| Minutes extracted | ||||||
| 0 | 2 | 5 | 10 | 20 | 30 | |
| Parameter | ||||||
| TnC (%) | 100 ± 4 | 77 ± 3 | 69 ± 8 | 40 ± 6 | 31 ± 2 | 18 ± 2 |
| Tension (%) | 100 ± 8 | 76 ± 8 | 40 ± 6 | 28 ± 6 | ||
| nH | 6.3 ± 0.5 | 3.3 ± 0.2 | 2.1 ± 0.01 | 2 ± 0.3 | ||
| pCa50 | 5.97 ± 0.02 | 5.65 ± 0.03 | 5.38 ± 0.05 | 5.21 ± 0.08 | ||
| Number fibres | 11 | 13 | 12 | 8 | ||
This data from Brandt, et al. [6] is somewhat confusing until one realises that the EDTA wash is also removing fsMyBP-C, almost certainly faster than the fsTnC. The result is the cooperativity, Hill coefficient, reaches that expected for solely the two Ca2+ binding sites on fsTnC being required for activation. This was 1984 as opposed to reference 7 which was later 1991 when this was realised by Moss [7].
Partial extraction of skeletal TnC in the presence of free Mg2+
By the addition of 1mM Mg2+ to the EDTA extraction procedure the loss of MyBP-C is minimised and this is ensured by only partial extraction of the fsTnC, Moss, et al. [26]. Note, Figure 7, only a small loss of cooperativity but considerable shift to lower pCa, higher [Ca2+] i.e. towards the Km of the Mg2+ dependent Ca2+ activation.
Total extraction of TnC from both skeletal and cardiac myofibrils with reversal
Morimoto and Ohtsuki changed the extraction in a similar way to Moss but with a different chelator. Endogenous troponin C in skinned psoas fibers and trabeculae was extracted by CDTA treatment until the preparations developed no Ca2+ activated tension or ATPase, i.e. complete extraction of TnC. The CDTA-treated preparations were either reconstituted or substituted with isolated fast-twitch skeletal or cardiac troponin C. When added back the Hill coefficients are as expected with partial MyBP-C extraction occurring as well. Can this be repeated with complete removal of the MyBP-C to give the skeletal and cardiac hill coefficients of 2 and 1 corresponding to fsTnC and cTnC binding alone? (Table 3).
| Table 3 | |||||
| Muscle preparation | pCa-tension | pCa ATPase | |||
| nH | pCa50 | nH | pCa50 | ||
| A) | Intact muscle | ||||
| Skeletal | 2.6 | 5.76 | 3.6 | 6.17 | |
| Cardiac | 1.8 | 5.7 | 2.0 | 5.75 | |
| B) | CDTA-treated muscle | ||||
| Skeletal | |||||
| + skeletal TnC | 2.7 | 5.85 | 3.0 | 5.94 | |
| + cardiac TnC | 1.4 | 5.37 | 1.5 | 5.50 | |
| Cardiac | |||||
| + skeletal TnC | 2.7 | 5.11 | 3.2 | 5.39 | |
| + cardiac TnC | 1.5 | 5.42 | 1.7 | 5.74 | |
| Similar extraction results were obtained by Zot and Potter [28]. | |||||
Similar extraction results were obtained by Zot and Potter [28].
The extraction of cMyBP-C from cardiac myofibrils
I now return to the data of Hofmann and their studies with partial extraction of subunits of the sarcomere [7]. In this first of their studies the extraction was limited to removal of solely MyBP-C by the addition of TnC to the extraction ensuring it remains in the myofibril. Other damage to the system was minimised by limiting the degree of extraction figure 8. As the extractions were only partial, unlike the unintentional loss of MyBP-C by Brandt, et al. [6] the precise values of the Hill coefficients are not relevant, so they are omitted, although follow the expected. The pCa dependency slopes and shifts are good enough indicators. In this exercise one can easily see the loss of Ca2+ cooperativity with a large shift toward the Km for TnC.
The process is reversible, Figure 9. Therefore the effect is due to cMyBP-C.
The extraction of fsMyBP-C from skeletal myofibrils (psoas fibres)
The same exercise as above was performed on skeletal muscle myofibrils figure 10. The authors explain difficulties in obtaining all the data on reversability. However, although the results are less spectacular than those from cardiac myofibrils the same effects are observed i.e. a pCa50 shift to lower [Ca2+] and decrease in the slope.
The extraction process was again reversible figure 11.
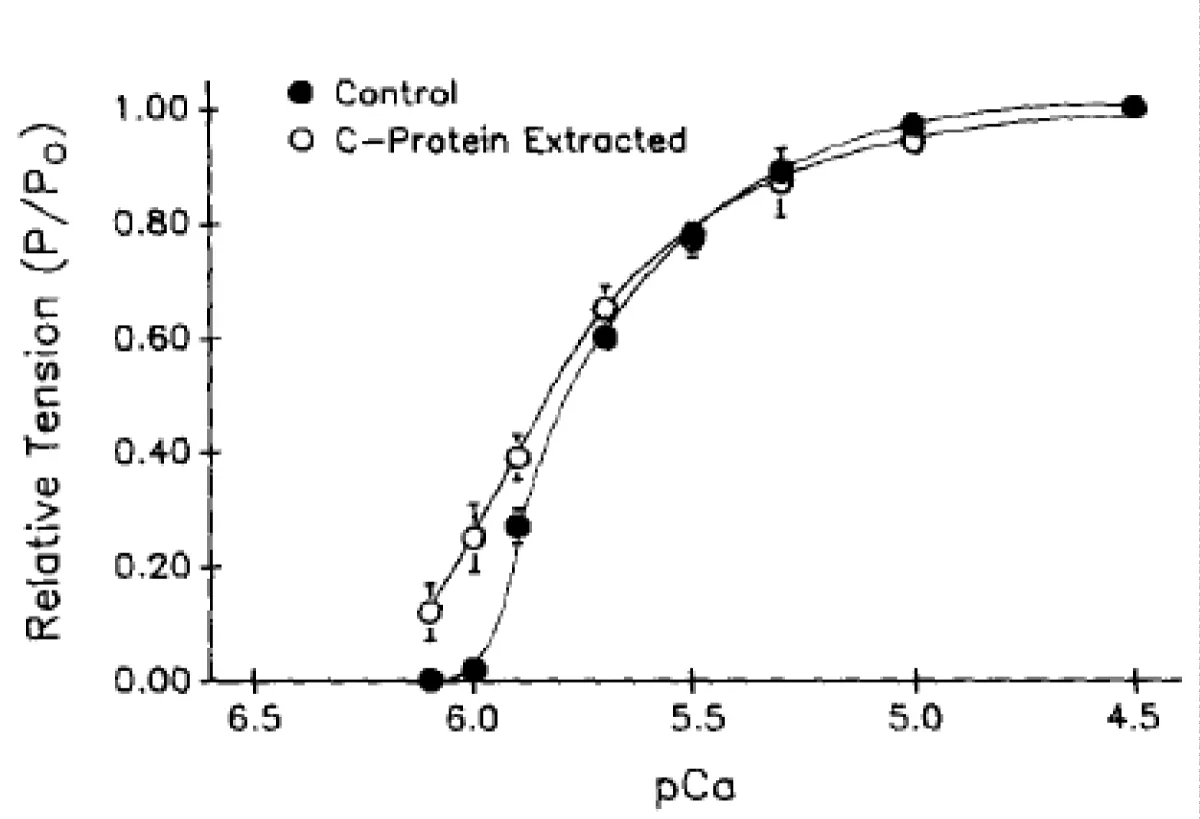
Figure 10: The effects of fsMyBP-C partial extraction on tension developed by rabbit psoas fibers, cumulative data.
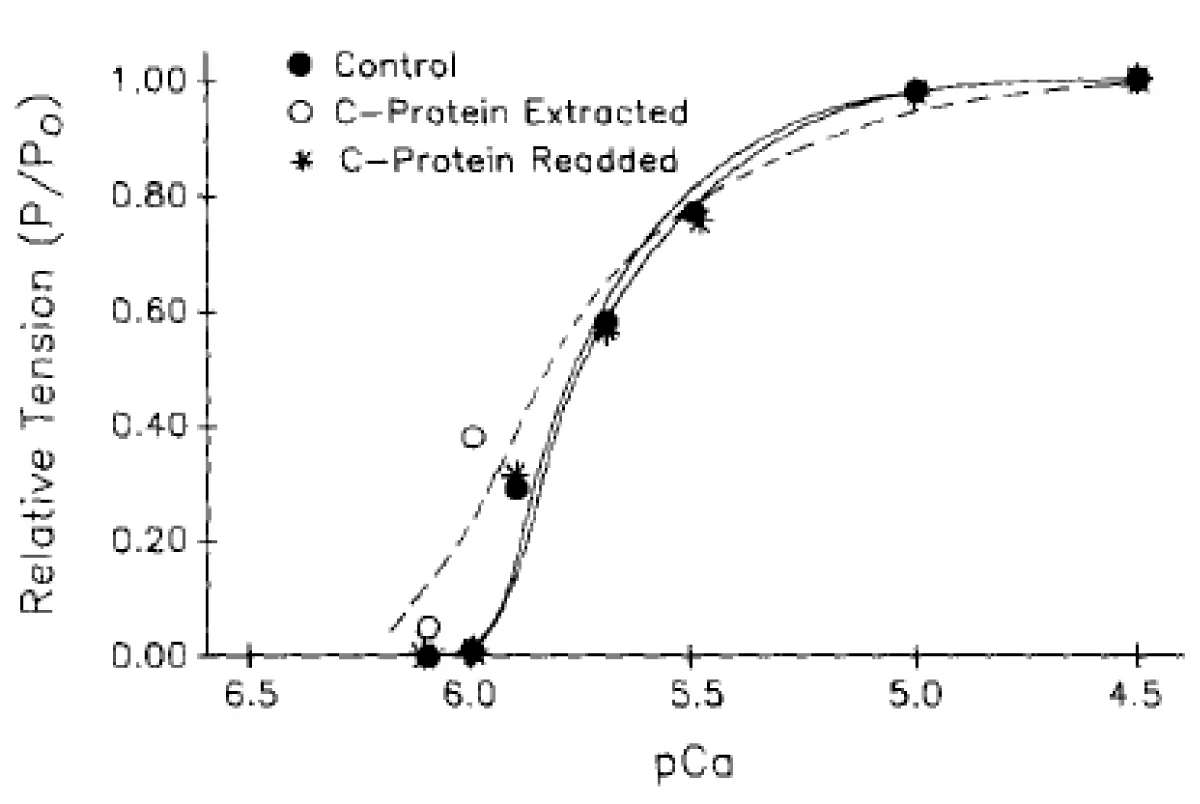
Figure 11: The effects of fsMyBP-C partial extraction on tension developed by rabbit psoas fibers, and to which fsMyBP-C was subsequently readded. After extraction, tension at pCa 4.5 was 0.99 of control Po; while after C-protein readdition, maximum tension was 0.90 of control Po.
The partial extraction of cTnC, and the subsequent extraction of cMyBP-C
I return to the cardiac data reported by Hofmann [7] to emphasise the strength of my earlier argument that there are two bindings of Ca2+ required for activation of muscle contraction, only one of which, on TnC, is measurable by binding of 45Ca2+. Figure 12 clearly shows the results of each extraction, both reduce the cooperativity and spectacularly give pCa shifts in opposite directions towards the Km of that Ca2+ activation protein not extracted. This is by far the best visual display of the duality of Ca2+ binding requirement for contraction that I have seen. It is absolutely clear that removal of the requirement for binding to the TnC the remaining control is at lower pCa, higher [Ca2+], and removal of the control by MyBP-C, the other control, is at higher pCa, lower [Ca2+] than the combination of both.
Figure 11: Data from a myocyte from which both cTnC and cMyBP-C were extracted: control (●), after extraction of cTnC (○), and after the subsequent extraction of cMyBP-C (▲). After extraction of cTnC, tension at pCa 4.5 was 0.40 of control Po; after extraction of cMyBP-C, tension was 0.37 control Po.
I have indicated that the basic functions of TnC and MyBP-C apply in both skeletal and cardiac systems, although modified, one can assume that the interactions described above also apply to TnI whether type 1,2 or 3. There are many conflicting reports in the literature on the function of MyBP-C including one where the title absolutely contradicts all current knowledge [16]. Of the other reports including one in 2018, e.g. [29-33] the key to the confusion is the unrelenting lack of acceptance of my 2001 paper with the unfounded dogma that protein activating ion binding sites must be measurable by radio-isotope binding.
Disruption of either cTnI or cMyBP-C, e.g. by mutation, can result in the shift of pCa50 to lower [Ca2+] and in the heart poor relaxation in diastole resulting in hypertrophic myopathy, but not in skeletal muscle. The main difference between the types of muscle are the rapid cycling of contraction in the heart, with guaranteed relaxation in diastole as opposed to the requirement for prolongation of contraction in the skeletal system and maintenance of developed tension for a period of time. These differences originate especially in the subunits of the sarcomere described here. In cardiac the Km for the Mg2+ dependant Ca2+ activation is at considerably higher [Ca2+] than that of the cTnC. This is not so for skeletal muscle. The above reported binding of the N-terminal MyBP-C to actin has resulted in the observation of isoforms of skeletal MyBP-C binding differently [34,35] and tune the molecular contractility of divergent skeletal muscle systems. I quote, “MyBP-C modulates muscle contractility, presumably through its N terminus extending from the thick filament and interacting with either the myosin head region and/or the actin thin filament. There is a further function of MyBP-C in that it binds creatine kinase ensuring that it is in the location of the cross-bridge ATPase as is MyBP-C and ensures the rapid replacement of the ADP and Pi produced with relaxing MgATP, [36]. One can only hope that the biochemical function of MyBP-C that I have described gets recognised soon and the research flourish.
All reproduced figures are in the public domain and the originals cited.
- Smith GA, Vandenberg JI, Freestone NS, Dixon HBF. The effect of Mg2+ on cardiac muscle function: Is CaATP the substrate for priming myofibril cross-bridge formation and Ca2+ reuptake by the sarcoplasmic reticulum? Biochem J. 2001; 354: 539-551. PubMed: https://pubmed.ncbi.nlm.nih.gov/11237858/
- Smith GA. Calcium, actomyosin kinetics, myosin binding protein-c and hypertrophic cardiomyopathy. J Integr Cardiol. 2019; 5: 1-2.
- Smith GA. The Mechanisms of the Frank-Starling Law and Familial Cardiomyopathy are Different. The Function of Myosin Binding Protein-C is Retained on Myocyte Length Increase and Force Generated is Kinase controlled. J Integr Cardiol. 2019; 5: 1-3.
- Smith GA. Angiotensin II type 1 receptor and the activation of Myosin Light-Chain Kinase and Protein Kinase C-βII: Mini Review. J Cardiol Cardiovasc Med. 2020; 5: 024-028. https://www.cardiologymedjournal.com/journals/jccm/jccm-aid1081.php
- Smith GA. The Mechanisms of Cardiac Myopathies, a kinetics approach. Leading review. J Cardiol Cardiovasc Med. 2020; 5: 141-152. https://www.cardiologymedjournal.com/journals/jccm/jccm-aid1101.php
- Brandt PW, Diamond MS, Gluck B, Kawai M, Schachat F. Molecular Basis of Cooperativity in Vertebrate Muscle Thin Filaments Carlsberg Res. Commun. 1984; 49: 155-167.
- Hofmann PA, Hartzell HC, Moss RL. Alterations in Ca2+ sensitive tension due to partial extraction of C-protein from rat skinned cardiac myocytes and rabbit skeletal muscle fibers. J Gen Physiol. 1991; 97: 1141-1163. PubMed: https://pubmed.ncbi.nlm.nih.gov/1678777/
- Katrukha IA. Human Cardiac Troponin Complex. Structure and Functions. Biochemistry (Moscow). 2013; 78: 1447-14650. PubMed: https://pubmed.ncbi.nlm.nih.gov/24490734/
- Layland J, Solaro RJ, Shah AM. Regulation of cardiac contractile function by troponin I phosphorylation. Cardiovascular Res. 2005; 66: 12–21. PubMed: https://pubmed.ncbi.nlm.nih.gov/15769444/
- Westfall MV, Lee AM, Robinson DA. Differential Contribution of Troponin I Phosphorylation Sites to the Endothelin-modulated Contractile Response. J Biol Chem. 2005; 280: 41324–41331. PubMed: https://pubmed.ncbi.nlm.nih.gov/16236710/
- Sheng JJ, Jin JP. TNNI1, TNNI2 and TNNI3: Evolution, regulation, and protein structure–function relationships. Gene. 2016; 576: 385-394. PubMed: https://pubmed.ncbi.nlm.nih.gov/26526134/
- Robinson P, Lipscomb S, Preston LC, Altin E, Watkins H, et al. Mutations in fast skeletal troponin I, troponin T, and beta-tropomyosin that cause distal arthrogryposis all increase contractile function. FASEB J. 2007; 21: 896–905. PubMed: https://pubmed.ncbi.nlm.nih.gov/17194691/
- Seidman JG, Seidman C. The genetic basis for cardiomyopathy: from mutation identification to mechanistic paradigms. Cell. 2001; 104: 557–567. PubMed: https://pubmed.ncbi.nlm.nih.gov/11239412/
- Curila K, Benesova L, Penicka M, Minarik M, Zemanek D, et al. Spectrum and clinical manifestations of mutations in genes responsible for hypertrophic cardiomyopathy. Acta Cardiol. 2012; 67: 23–29. PubMed: https://pubmed.ncbi.nlm.nih.gov/22455086/
- Heling LWHJ, Geeves MA, Kad NM. MyBP-C: one protein to govern them all. J. Muscle Res. and Cell Motility. 2020; 41: 91–101.
- Lin B, Govindan S, Lee K, Zhao P, Renzhi Han R, et al. Cardiac Myosin Binding Protein-C Plays No Regulatory Role in Skeletal Muscle Structure and Function. PLoS One. 2013; 8: e69671. PubMed: https://pubmed.ncbi.nlm.nih.gov/23936073/
- Lee RS. Effect of the Ca2+ Binding Properties of Troponin C On Skeletal and Cardiac Muscle Force Development. Thesis the Ohio State University. 2010.
- Potter JD, Gergely J. J BiolChem. 1975; 250: 4628-4633.
- Kretsinger RH. CRC Crit Rev Biochem. 1980; 8: 119-174.
- Sia SK, Li MX, Spyracopoulos L, Gagne SM, Liu W, et al. J Biol Chem. 1997; 272: 18216-18221.
- Brandt PW, Cox RN, Kawai M. Can the binding of Ca2+ to two regulatory sites on troponin-C determine the steep pCa/tension relationship of skeletal muscle? Proc Natl Acad Sci. 1980; 77: 4717-4720. PubMed: https://pubmed.ncbi.nlm.nih.gov/6933518/
- Kampourakis T, Yan Z, Gautel M, Sun YB, Irving M. Myosin binding protein-C activates thin filaments and inhibits thick filaments in heart muscle cells PNAS. 2014; 111: 18763–18768. PubMed: https://pubmed.ncbi.nlm.nih.gov/25512492/
- Flashman E, Watkins H, Redwood C. Localization of the binding site of the C-terminal domain of cardiac myosin-binding protein-C on the myosin rod. Biochem J. 2007; 401: 97–102. PubMed: https://pubmed.ncbi.nlm.nih.gov/16918501/
- van Dijk SJ. Bezold KL, Harris SP. Earning Stripes: Myosin Binding Protein-C Interactions with Actin. Pflugers Arch. 2014; 466: 445–450. PubMed: https://pubmed.ncbi.nlm.nih.gov/24442149/
- Lu Y, Kwan AH, Trewhella J, Jeffries CM. Communication: The C0C1 Fragment of Human Cardiac Myosin Binding Protein C Has Common Binding Determinants for Both Actin and Myosin. J Mol Biol. 2011; 413: 908–913. PubMed: https://pubmed.ncbi.nlm.nih.gov/21978665/
- Moss RL, Giulian GG, Greaser ML. The effects of partial extraction of TnC upon the tension-pCa relationship in rabbit skinned skeletal muscle fibers. J Gen Physiol. 1985; 86: 585–600. PubMed: https://pubmed.ncbi.nlm.nih.gov/4056736/
- Morimoto S, Ohtsuki I. Role of Troponin C in Determining the Ca2+ -Sensitivity and Cooperativity of the Tension Development in Rabbit Skeletal and Cardiac Muscles. J Biochem. 1994; 115: 144-146.
- Zot HG, Potter JD. A structural role for the Ca2+-Mg2+ sites on troponin C in the regulation of muscle contraction. Preparation and properties of troponin C depleted myofibrils. J Biol Chem. 1982; 257: 7678-7683. PubMed: https://pubmed.ncbi.nlm.nih.gov/6211445/
- Pohlmann L, Kröger I, Vignier N, Schlossarek S, Krämer E, et al. Cardiac Myosin-Binding Protein C Is Required for Complete Relaxation in Intact Myocytes. Circ Res. 2007; 101: 928-938. PubMed: https://pubmed.ncbi.nlm.nih.gov/17823372/
- Lecarpentier Y, Vignier N, Oliviero P, Guellich A, Carrier L, et al. Cardiac Myosin-Binding Protein C Modulates the Tuning of the Molecular Motor in the Heart. Biophysical J. 2008; 95: 720–728. PubMed: https://pubmed.ncbi.nlm.nih.gov/18375505/
- Razumova MV, Bezold KL, Tu AY, Regnier M, Harris SP. Contribution of the Myosin Binding Protein C Motif to Functional Effects in Permeabilized Rat Trabeculae. J Gen Physiol. 2008; 132: 575–585. PubMed: https://pubmed.ncbi.nlm.nih.gov/18955596/
- Lin BL, Li A, Mun JY, Previs MJ, Previs SB, et al. Skeletal myosin binding protein-C isoforms regulate thin filament activity in a Ca2+-dependent manner. Nature, Scientific Reports. 2018; 8: 2604. PubMed: https://pubmed.ncbi.nlm.nih.gov/29422607/
- Oakley CE, Chamoun J, Brown LJ, Hamblya BD. Binding protein-C: Enigmatic regulator of cardiac contraction. Int J Biochem Cell Biol. 2007; 39: 2161–2166. PubMed: https://pubmed.ncbi.nlm.nih.gov/17320463/
- Orlova A, Galkin VE, Cy MJ, Jeffries Cy MJ, Egelman EH, et al. The N-terminal Domains of Myosin Binding Protein C Can Bind Polymorphically to F-Actin. J Mol Biol. 2011; 412: 379–386. PubMed: https://pubmed.ncbi.nlm.nih.gov/21821050/
- Li A. Nelson SR, Rahmanseresht S, Braet F, Cornachione AS, et al. Skeletal MyBP-C isoforms tune the molecular contractility of divergent skeletal muscle systems. PNAS. 2019; 116: 21882–21892. PubMed: https://pubmed.ncbi.nlm.nih.gov/31591218/
- Chen Z, Zhao TJ, Li J, Gao YS, Meng FG, et al. Slow skeletal muscle myosin-binding protein-C (MyBPC1) mediates recruitment of muscle-type creatine kinase (CK) to myosin. Biochem J. 2011; 436: 437-445. PubMed: https://pubmed.ncbi.nlm.nih.gov/21426302/