
More Information
Submitted: February 07, 2021 | Approved: March 09, 2021 | Published: March 12, 2021
How to cite this article: Smith GA. Muscle growth and control of production of sarcomere components. J Cardiol Cardiovasc Med. 2021; 6: 019-022.
DOI: 10.29328/journal.jccm.1001111
ORCiD: orcid.org/0000-0002-3959-3523
Copyright License: © 2021 Smith GA. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Muscle growth and control of production of sarcomere components
Gerry A Smith*
Retired, Department of Biochemistry, University of Cambridge, Tennis Court Rd. Cambridge CB2 1QW, UK
*Address for Correspondence: Gerry A Smith, Retired, Department of Biochemistry, University of Cambridge, Tennis Court Rd. Cambridge CB2 1QW, UK, Tel: +44 1223 515394; Email: [email protected]
This presentation gives a description of the muscle and sarcomere followed by the main content summarized.
Here I contrast the skeletal and cardiac muscle in terms of the control muscle growth and of sarcomere component synthesis. The differences are major and reflect the long term needs of the two systems. With the skeletal system there is growth of both the number of myocytes and the sarcomere components within them dependent on demand made of the muscle. Unlike skeletal muscles the normal adult heart is greatly restricted in size, number of myocytes and their content of contractile proteins, i.e. there is little change on demand. Over time proteins get damaged or decay and for the normal heart this implies a strictly controlled maintenance synthesis of sarcomere components. From the studies of abnormal, mutated systems there is one thing inherent to and more pronounced in cardiac muscle, the FrankStarling Law of the Heart derived from the angiotensin ii type 1 receptor that my studies indicate is central to the control of sarcomere component synthesis.
The muscle and sarcomere construction
The muscle is made up of long cells called myocytes arranged side by side to give force in one direction with their length. They contain the contractile apparatus which is referred to as the sarcomere. Activation is via depolarization allowing Ca2+ to rise in the cytosol, in skeletal this is driven by nervous control and can be sustained, in heart muscle it is a cyclical event giving the recognized heart or pulse rate.
The contractile apparatus, the sarcomere, comprises two overlapping filaments in the relaxed state. the thick filament (optically darker A band) and the thin filament (optically lighter I band) as shown in figure 1. Cross bridges form between the two bands and consume ATP as they slide together forcibly shortening the sarcomere.
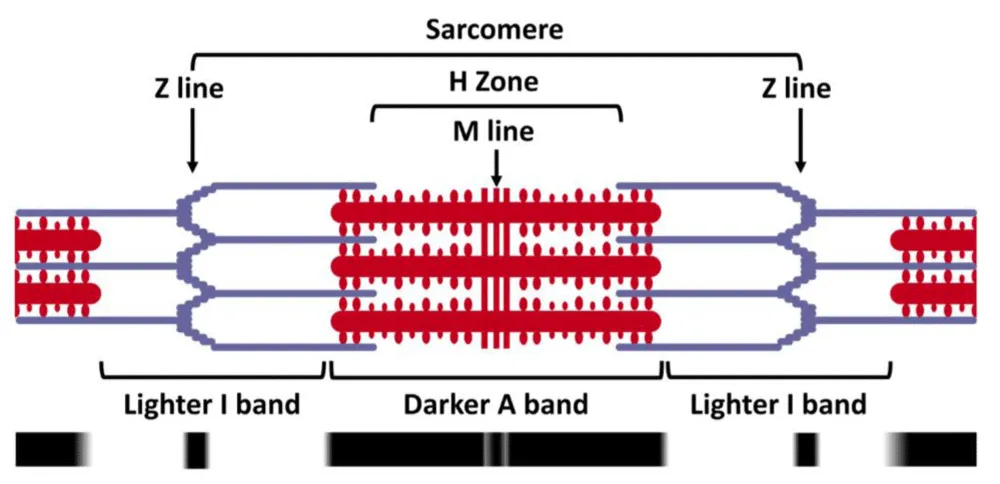
Figure 1:
The thin filament is of actin polymer and has bound to it various components, the key Ca2+ switch protein troponin-C (TnC), the control protein troponin-I (TnI) the protein that blocks cross-bridge forming to the action until displaced by TnI after TnC-Ca2+ binding.
There is also troponin-T (TnT) involved in tension transfer along the thin filament.
The thick filament comprises myosin polymer which has protruding towards the thin filament the myosin light chain (MLC) which when activated links to the actin filament using ATP, the cross-bridge. The MLC binds the metal complex of the ATP. Among other things bound to the thick filament is what I describe as the critical control protein myosin binding protein-C (MyBP-C) whose biochemical function is to block MLC bound MgATP from being used as substrate for cross-bridge formation until the Mg2+ is replaced by Ca2+ on the MLC. Along with the filaments is the giant protein titin which incorporates the growth control elements, reporting by the tension detected by it.
Summary of content
The growth or absence of it in the two main muscle types is described followed by concentration on the cardiac muscle with particular consideration of the two main abnormalities, myopathies, found. The growth mis-control found within these abnormalities and with certain drug administrations is very informative on mechanisms if they are considered carefully. One drug, losartan and its copies, is of particular interest because incorrectly used can do damage. It blocks the angiotensin ii type 1 receptor which controls the Frank-Starling law and the background regeneration of the sarcomere. In doing so in non-hypertrophic cases it leads to a loss of sarcomere components and dilated myopathy, heart-failure. I then quote myself as a case of the misuse of this drug.
Skeletal muscle
For skeletal muscle the system is simple. When there is more neuromuscular stimulation then the muscle will grow in size and strength and for this there is obvious production of new myocytes and the required sarcomere components. If there is loss of neuromuscular stimulation the muscle wastes away as there is reduced production of the sarcomere components [1]. Considerable is known about the specifics of how the tension within the muscle filaments is involved in the control of this process. The tension gets transferred to the giant molecule titin which has a flexible portion which when stretched releases growth factors [2].
Cardiac muscle
This is where the difference in muscle type is most apparent. In the normal adult heart there is no change in size or number of muscle cells, myocytes [3]. The heart stays constant in this regard, however over a lifetime there will be degeneration of the components of the sarcomere by shear wear and tear. In order to maintain this constancy within the normal heart there has to be a central control of the production of the sarcomere components.
In the abnormal heart, i.e. those affected by either catastrophic events, e.g. infarct, or abnormality from birth, i.e. genetic errors, the control system under discussion can break down. There can be excessive production of sarcomere components and incorporation of them in the existing myocytes, leading to hypertrophic myopathy, excessive filaments in the myocyte [4]. The converse situation is another common result of mutations, i.e. failure to produce enough sarcomere components leading to loss of muscle strength, dilated cardiomyopathy [4-6]. These events are observed as only partial as extremes result in death. A direct comparison of skeletal and cardiac muscle particularly from the kinetic approach is given by me [7].
Hypertrophic cardiomyopathy
The mechanisms behind the former of these conditions, hypertrophy, I have dealt with in great detail [4-6]. This has required the overturning of a long held belief that the substrate of the normal cross-bridge ATPase is MgATP and showing that it is actually CaATP [4-6]. Crossing this bridge also generated the first biochemical function of myosin binding proteinC (MyBP-C) which is not simply structural but blocks the use of MgATP as substrate requiring that the Mg2+ is swapped for Ca2+ for activity. This is the second, cooperative Ca2+ that is well known in the cardiac kinetics, there is only one Ca2+ binding site measurable with 45Ca. The biochemical function of MyBP-C ensures that MgATP bound to the myosin light chain in diastole is the truly relaxed state [4,5]. When this goes wrong and MgATP is used, the Ca2+ cooperativity of the ATPase is lost, lowered Hill coefficient for Ca2+ activation, and the stimulation of contraction occurs at lower [Ca2+], approaching the Km of troponin-C for Ca2+, and relaxation in diastole is incomplete. This ‘going wrong’ gives chronic stress within the filaments and stretch of the elastic part of titin resulting in an increase of sarcomere component production, the result is hypertrophy. As expected this is most marked for mutations in MyBP-C and the other components it binds to. However the system is so integrated that mutations in almost all components can produce the reduction in Ca2+ cooperativity, Hill coefficient, leading to hypertrophy [6]. There are also a few cases of hypertrophy not showing loss of Ca2+ cooperativity which are presumed to be mutations in filament proteins leading to increased transmission of tension [6]. There are many publications showing that the use of angiotensin ii type 1 inhibitors are greatly beneficial in cases of ventricular hypertrophy in that they inhibit the progression of the hypertrophy, i.e. they inhibit the over-production of sarcomere components [8]. This I will return to later.
Dilated cardiomyopathy
Here the control of production of replacement sarcomere components breaks down and less than that needed for maintenance are produced. The resulting ventricle without proper full contraction apparatus becomes floppy and generally enlarged, dilated. There are many mutations of sarcomere constituents giving rise to dilated cardiomyopathy and as shown in virtually all cases there is no loss or increase in the Ca2+ cooperativity observed [6]. In many instances of familial dilated myopathy, there is also the noted absence of the effect of diastolic stretch on the strength of contraction [9], Frank-Starling Law of the heart [10]. As with hypertrophy there will be instances where there are changes in the efficiency of transmission of tension within the fibres, here leading to less effect on the elastic portion of titin resulting in reduced production of sarcomere components.
There is nothing that tells us the seat of generation of the signal for maintenance of component production. This must be within the cardiac muscle specifics and likely to have relationship to retention of the size of the myocyte, only when things go wrong in hypertrophic or dilated cardiomyopathy does one get too much or too little sarcomere in the myocyte. There is one outstanding candidate for control of this and that is the Angiotensin II type 1 receptor and its relationship to the Frank-Starling Law of the heart [9,11,12]. The increased systolic tension within the sarcomere that occurs when the myocyte is lengthened in diastole is a major candidate for control of component production. As I have restated from Abraham [9,11,12] losartan blocks the Frank-Starling Law which arises from activation of the angiotensin ii type 1 receptor by increase in length of the myocyte in the absence of ligand [12]. The control of myosin light chain kinase (MLCK) and Protein Kinase C-βII by the kinase inhibitor β-Arrestin and the kinase target proteins, the MLC and troponin-I at threonine 144 are in need of more in depth analysis.Angiotensin block on normal or dilated hearts
However much I search I find no definitive study of the effect of long-term suppression of the angiotensin ii type 1 receptor on the non-hypertrophic heart, these probably used but failed treatments are lost in the positive reporting of curing of hypertrophy, most referred for hypertension will be at minimum bordering on hypertrophy. In the light of the often and much heralded benefits angiotensin ii block, particularly with losartan, on ventricular hypertrophy by blocking sarcomere component production, I am extremely surprised not to be able to find reports of it causing dilated myopathy in the normal heart. These two observations are not compatible. There are confusing reports that the vastly different angiotensin ii block and ACE inhibitors do like things but never clear what and they are generally grouped together, this sort of mix up is unwelcome e.g. [13]. There is a long overdue echocardiogram study required into the relationship of specific hypertension drug type usage and ventricular dilation in non-hypertrophic patients. The truth is however revealed in statements released by pharmaceutical companies and I quote “In patients with heart failure, with or without renal impairment, there is - as with other medicinal products acting on the renin-angiotensin system - a risk of severe arterial hypotension.” and “There is evidence that the concomitant use of ACE-inhibitors, angiotensin II receptor blockers or aliskiren increases the risk of hypotension, Heart failure” [14]. My own interpretation of these statements is for hypotension and heart failure read dilated cardiomyopathy.
I believe that I am one directly affected by this till now not fully understood science of the heart. After aortic valve malfunction causing infarct I had a metal aortic valve fitted and more than ten years of successful old styled treatment for mild hypertension along with of course warfarin. A couple of years back with a rash complication I was switched to losartan.
I now have serious dilation of the heart, listed as heart failure, extreme hypotension resting BP 100-105/50-60 with heart rate 75-85, treated with bisoprolol. Added to this is the complication of the implant of a pacemaker for a break in conductance. This last issue might indicate that ion channels are also under the same control as the sarcomere components as one would expect [15] and mine are now probably much lower in number. I have now stopped the beta-blocker bisoprolol, and after an initial heart rate rise to ca 100 bpm (fast from limited depolarization) and is now reducing around 85-90 bpm (greater degree of depolarisation) and a much more stable and sensible BP of around or a bit below 120/65.
Notes added in proof
These display the importance of the angiotensin ii type 1 receptor, the MLCK and the Frank -Starling Law in the generation and maintenance of sarcomere.
The earliest of these is by Takashima, et al. [16] who have shown clearly in cultured cardiomyocytes, knockdown of cardiac-MLCK decreased MLC2v phosphorylation and impaired epinephrine-induced activation of sarcomere reassembly. Knockdown of z–cardiacMLCK expression in zebra fish resulted in dilated cardiac ventricles and immature sarcomere structures.
These studies were followed much later in 2016 by Irving, et al. [17] who used phosphorylation of the myosin regulatory light chain (cRLC) by the cMLCK to show that cRLC phosphorylation enhanced active force and its calcium sensitivity and altered thick filament structure as reported by bifunctional rhodamine probes on the cRLC: the myosin head domains became more perpendicular to the filament axis. The effects of cRLC were mimicked by increasing sarcomere length i.e The Frank-Starling Law.
Kasahara, et al. [18] using knockdown of cMLCK in cardiomyocytes that resulted in reduced cardiac contractility and sarcomere disorganization, hypothesized that acute reduction of cMLCK may be causative for reduced contractility and cardiomyocyte remodelling during the transition from compensated to decompensated cardiac hypertrophy.
It is very clear that in the normal heart the initiation the synthesis of the sarcomere components required for maintenance resides in the angiotensin ii type 1 receptor and probably in particular the increase in contraction on diastolic myocyte lengthening, the Frank-Starling Law. The mechanism for limiting the production of components is still not obvious but may also lie within the same restriction of more synthesis being limited to ligand free activation of the angiotensin receptor. Turnover as a result of this restriction may be just sufficient for renewal. On the overall perspective presented here there has to be a block on sarcomere component production from increased kinase A or C activation. The myosin light chain kinase, essential for length activation, is not activated but controlled by the kinase inhibitor β-Arrestin. The β-Arrestin is relocated by action of the angiotensin ii type 1 receptor.
The sarcomere picture courtesy of Shutterstock, Inc. | 350 Fifth Avenue, 21st Floor | New York, NY 10118 | USA.
There are no conflicts of interest and no funding oof this work.
- Morgan J, Partridge T. Skeletal muscle in health and disease: Disease Models & Mechanisms. 2020; 13: dmm042192. PubMed: https://pubmed.ncbi.nlm.nih.gov/32066552/
- Linke WA, Hamdani N. Gigantic Business: Titin Properties and Function through Thick and Thin. Circ Res. 2014; 114:1052-1068. PubMed: https://pubmed.ncbi.nlm.nih.gov/24625729/
- Günthel M, Barnett P, Christoffels VM. Review; Development, Proliferation, and Growth of the Mammalian Heart. Mol Ther. 2018; 26: 1599-1609. PubMed: https://pubmed.ncbi.nlm.nih.gov/29929790/
- Smith GA. Calcium, actomyosin kinetics, myosin binding protein-c and hypertrophic cardiomyopathy. J Integr Cardiol. 2019; 5: 1-2.
- Smith GA, Vandenberg JI, Freestone NS, Dixon HBF. The effect of Mg2+ on cardiac muscle function: Is CaATP the substrate for priming myofibril cross-bridge formation and Ca2+ reuptake by the sarcoplasmic reticulum? Biochem J. 2001; 354: 539-551. PubMed: https://pubmed.ncbi.nlm.nih.gov/11237858/
- Smith GA. The Mechanisms of Cardiac Myopathies, a kinetics approach. Leading review. J Cardiol Cardiovasc Med. 2020; 5: 141-152. PubMed: https://www.cardiologymedjournal.com/journals/jccm/jccm-aid1101.php
- Smith GA. New Insights from Cardiac Muscle Applied to Skeletal Muscle. J Cardiol Cardiovasc Med. 2021; 6: 007-013. PubMed: https://www.cardiologymedjournal.com/journals/jccm/jccm-aid1109.php
- Doggrell SA. Losartan for LIFE in hypertension with left ventricular hypertrophy? Expert Opin Pharmacother. 2003; 4: 115-118. PubMed: https://pubmed.ncbi.nlm.nih.gov/12517248/
- Smith GA. The Mechanisms of the Frank-Starling Law and Familial Cardiomyopathy are Different. The Function of Myosin Binding Protein-C is Retained on Myocyte Length Increase and Force Generated is Kinase controlled. J Integr Cardiol. 2019; 5: 13.
- Katz AM. Ernest Henry Starling, His Predecessors, and the "Law of the Heart". Circulation. 2002; 106: 2986–2992. PubMed: https://pubmed.ncbi.nlm.nih.gov/12460884/
- Abraham DM, Davis RT, Warren CM, Mao L, Wolska BM, et al. β-Arrestin mediates the Frank–Starling mechanism of cardiac contractility. PNAS. 2016; 113: 14426–14431. PubMed: https://pubmed.ncbi.nlm.nih.gov/27911784/
- Smith GA. Angiotensin II type 1 receptor and the activation of Myosin LightChain Kinase and Protein Kinase C-βII: Mini Review. J Cardiol Cardiovasc Med. 2020; 5: 024-028. PubMed: https://www.cardiologymedjournal.com/journals/jccm/jccm-aid1081.php
- Hoshikawa E, Matsumura Y, Kubo T, Okawa M, Yamasaki N, et al. Effect of Left Ventricular Reverse Remodeling on Long-TermPrognosis After Therapy With Angiotensin-Converting Enzyme Inhibitors or Angiotensin II Receptor Blockers and β Blockers in Patients With Idiopathic Dilated Cardiomyopathy. Am J Cardiol. 2011; 107: 1065–1070. PubMed: https://pubmed.ncbi.nlm.nih.gov/21296328/
- Losartan Potassium 25 mg film coated tablets, Last updated on emc: 13 Mar 2020. Datapharm. The electronic medicines compendium (emc). PubMed: https://www.medicines.org.uk/emc/product/8735/smpc
- Goette A, Lendeckel U. Electrophysiological effects of angiotensin II. Part I: signal transduction and basic electrophysiological mechanisms. Europace. 2008; 10: 238– 241. PubMed: https://pubmed.ncbi.nlm.nih.gov/18256129/
- Seguchi O, Takashima S, Yamazaki S, Asakura M, Asano Y, et al. A cardiac myosin light chain kinase regulates sarcomere assembly in the vertebrate heart. J Clin Invest. 2007; 117: 2812–2824. PubMed: https://pubmed.ncbi.nlm.nih.gov/17885681/
- Kampourakis T, Suna YB. Irving M. Myosin light chain phosphorylation enhances contraction of heart muscle via structural changes in both thick and thin filaments. PNAS. 2016; E3039–E3047. PubMed: https://pubmed.ncbi.nlm.nih.gov/27162358/
- Massengill MT, Ashrafm HM, Chowdhury RR, Chrzanowski SM, Kar J, et al. Acute heart failure with cardiomyocyte atrophy induced in adult mice by ablation of cardiac myosin light chain kinase. Cardiovasc Res. 2016; 111: 34–43. PubMed: https://pubmed.ncbi.nlm.nih.gov/27025239/