
More Information
Submitted: February 26, 2020 | Approved: December 26, 2022 | Published: December 27, 2022
How to cite this article: Christian MGR, Koudougou KJ, Arthur STA, Théodore B, Laurence B, et al. Double aortic dissection in a patient with Marfan disease. A case report. J Cardiol Cardiovasc Med. 2022; 7: 115-118.
DOI: 10.29328/journal.jccm.1001145
Copyright License: © 2022 Christian MGR, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Double aortic dissection in a patient with Marfan disease. A case report
Millogo Georges Rosa Christian*, Kologo Jonas Koudougou, Seghda Taryètba André Arthur, Boro Théodore, Benon Laurence, Samadoulougou André K and Zabsonré Patrice
Cardiology Department, University Hospital Yalgado Ouedraogo, Burkina Faso
*Address for Correspondence: Millogo Georges Rosa Christian, Cardiology Department, University Hospital Yalgado Ouedraogo, Burkina Faso, Email: [email protected]
Marfan syndrome is a dominant autosomal genetic disease of the connective tissue, leading to various complications. Cardiovascular complications are the main ones, with dilation of the aorta and aortic dissection which is the main cause of death. Double dissection with different sites of departure is scarcely reported by the literature. We reported the case of a 22-year’s-old young patient admitted for acute chest and abdominal pains, and cardiogenic shock. Investigations reported a double dissection of the aorta with a recent one of type A and an old one of type B. Ghent diagnostic criteria were used to allow the diagnosis of Marfan disease. Surgical management corrected successfully the type A dissection.
Early diagnostic and adequate management of aortic complications can reduce the high mortality rate of patients with Marfan syndrome.
Described by Antoine Marfan in 1896, Marfan syndrome is an abnormality of the connective tissue usually due to a mutation in the fibrillin-1 (FBN1) gene on chromosome 15, which determines fibrillin structure [1,2]. Scarcely, it can be a mutation in the gene encoding for the TGF-beta receptor [3]. Marfan syndrome is a dominant autosomal genetic disorder [3,4]. The diagnosis is based on the 2010 revised Ghent criteria including aortic root dilation, ectopic lens, and genetic mutation [4,5]. The prevalence in the general population is 2 - 3 per 10000 inhabitants [6-8]. Vital prognosis is essentially determined by cardiovascular damages, mainly made of mitral and aortic lesions. Gradual dilation of the aortic root leading to type A aortic dissection and aortic rupture are the main causes of death. In-hospital mortality from acute aortic dissection is about 15% to 25% [9].
In Marfan patients, acute aortic dissection occurs more on the dilated aortic root, or the ascending aorta just after the origin of the coronary arteries (type A). The dissection extends most of the time from the aortic arch to the abdominal aorta. A small percentage of these aortic dissections affect only the descending and abdominal aorta (type B). The dissection is usually of type A, rarely of type B and exceptionally associates both types [10].
We report here a case of type A and B dissection of an aortic aneurysm in a patient with Marfan syndrome, followed by a review of the literature.
Mr. K W, 22 years old, was admitted on February 24th, 2013 to the cardiology department for sudden onset of orthopnea with severe chest and abdominal pains, and cough with bloody sputum. Past medical history reveals the progressive onset of dyspnea for about a month; thereafter the patient reported sudden severe chest and abdominal pains described as a tear radiating down and backward. A cough with hemoptysis was also reported. After the failure of ambulatory treatment and the onset of NYHA class IV dyspnea, the patient was referred to the cardiology department for better management.
On admission, the general condition and the consciousness of the patient were impaired. The hemodynamic condition was unstable with a blood pressure of 100/50 mmHg on both arms; heart beats were regular with a rate of 110 beats per minute; the patient was breathless in the supine position, jugular veins were distended and neck vessels were hyper-pulsatile. Physical examination noticed cold extremities and discreet edema of the lower limbs; weight was 72 kg for a height of 205 cm (body surface was 2.10 m²). There was a general skeletal dysmorphia with a Marfan-like frame, a protrusion of the acetabulum, a pectus excavatum, and a span-over-height ratio of 1.2. Cardiovascular examination reported chest and abdominal pain syndrome, congestive heart failure, and aortic diastolic murmur of 4/6. The cardiovascular risk factor was active smoking (2.1 pack-years).
Laboratory examination reported a hemoglobin level of 12.2 g/dl with normal platelets and white blood cells count; Kidney function test and blood electrolytes were normal; the cycle of serum troponin and creatinine-phosphokinase levels were normal; HIV retroviral serology was negative. Chest x-ray reported a cardiomegaly (cardiothoracic index of 0.6) with pulmonary edema and an enlarged upper right arch of the mediastinum (Figure 1). A transthoracic electrocardiogram recorded regular sinus tachycardia (110 cycles per minute), left anterior hemiblock and poor progression of R waves in the anterior leads (Figure 2).
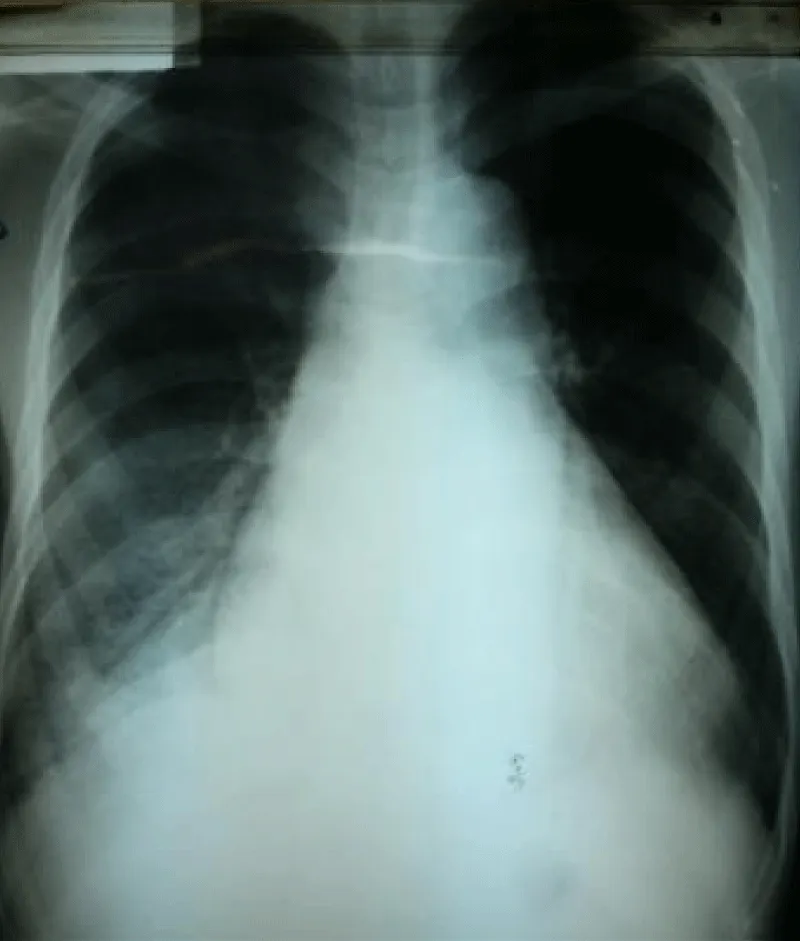
Figure 1: Frontal chest x-ray showing global cardiomegaly with signs of cardiac lung and enlargement of the upper mediastinum with overhang of the right upper arch.
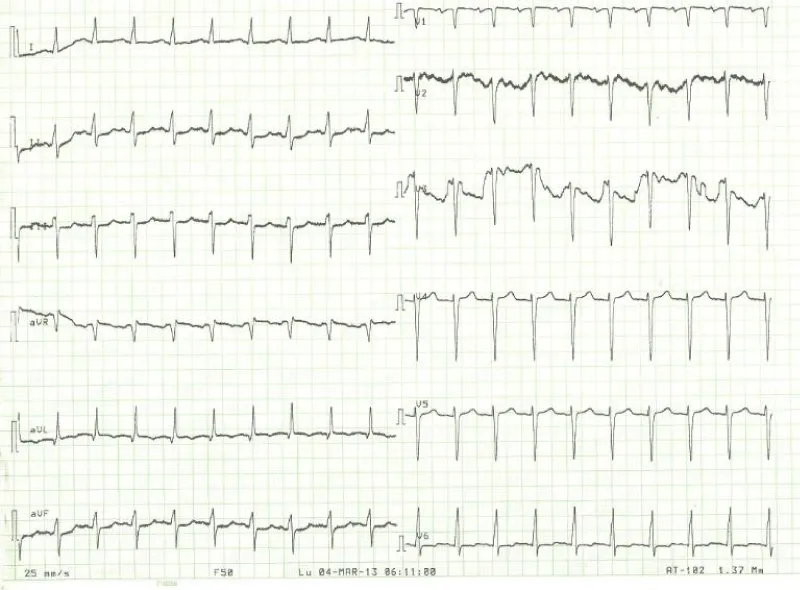
Figure 2: 12-leads electrocardiogram with sinus tachycardia, no R-wave progression in the antero-septal.
Transthoracic echocardiography (Figures 3,4) found a dilated aortic root (52 mm) with an ultrasound image of the intimal flap, and significant bi-ventricular dilation (left ventricular end-diastolic diameter of 72 mm). Systolic functions of both ventricles were severely impaired (LVEF of 24% and TAPSE of 10 mm). There was severe aortic regurgitation with a regurgitant orifice area of 35 mm². Dissection of an aortic aneurysm on dilated cardiomyopathy was diagnosed and confirmed by aortic CT scan (Figure 5); it was a type A aortic dissection with very severe dilation of the aortic root at the origin of the first branch of the supra-aortic trunks. A Type B dissection coexisted along, with an aortic dilation beginning far from the origin of the last branch of the supra-aortic trunks up to the iliac bifurcation. The Marfan disease was diagnosed despite the absence of genetic testing and a family history of the syndrome. An ophthalmological assessment could not be carried out. Medical treatment with diuretics, vasopressin amines, and analgesics was started and enabled slight improvement. The patient was later referred for surgical management through the financial support of a philanthropic sponsor. He was operated on March 7th, 2013 under cardiopulmonary bypass, using the Tirone David procedure. During surgery, the impression of two separate aortic dissections was confirmed: the former one of type B which previously went unnoticed, and the recent one of type A which determined the patient current clinical condition. After surgery, congestive heart failure was persistent, but during follow-up in the outpatient unit, the clinical condition remained stable under diuretic, ACE inhibitor, and oral anticoagulation. Unfortunately, the patient died four years later in 2017, due to poor medical follow-up.
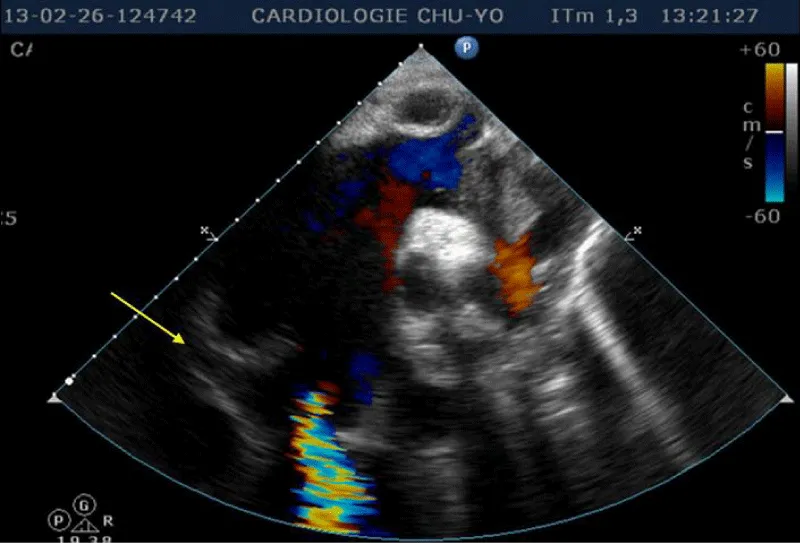
Figure 3: Transthoracic heart ultrasound suprasternal incidence: note the aneurysmal dilatation of the ascending aorta, aortic leakage, presence of an intimal tear and intimal flap (arrow).
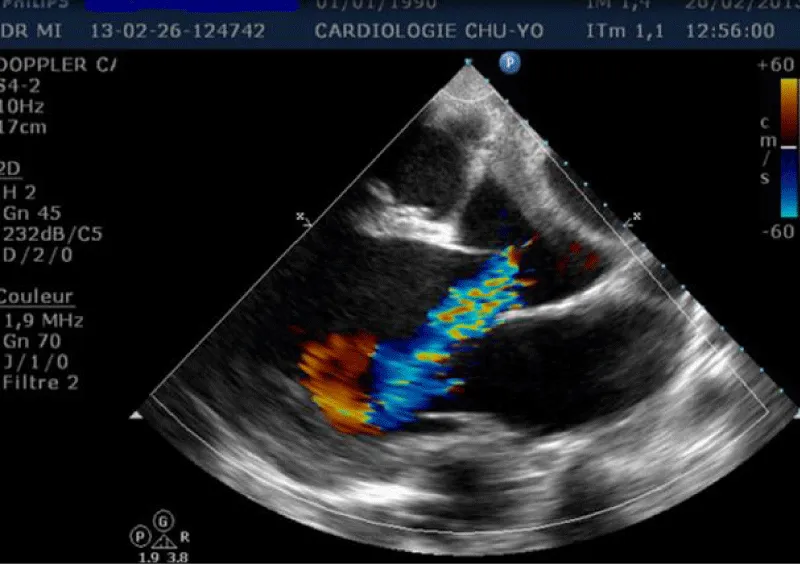
Figure 4: Transthoracic heart ultrasound para-sternal long axis incidence: dilatation of the ascending aorta and aortic leakage directed toward the large mitral valve.
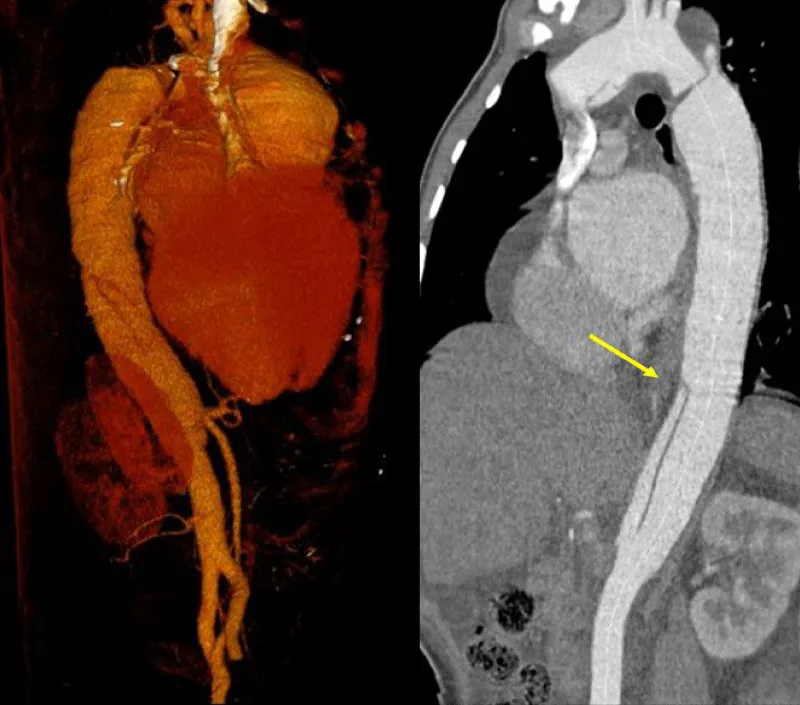
Figure 5: Aortic CT angiography: note the two levels of the intimal tear. The first is located in the ascending aorta with aneurysmal dilatation of this aortic portion and the second dissection originating distally on the abdominal aorta up to the iliac bifurcation.
Marfan syndrome is a rare and severe disease in our African context. It is one of the main causes of aortic dissection in young patients under the age of 40, according to the International Registry of Aortic Dissection IRAD [11]. The diagnosis could be missed in our context, in the absence of genetic testing. More recently the diagnosis was simplified by the 2010 revised Ghent criteria, including skeletal, ocular, cardiovascular, pulmonary, neurological, and cutaneous manifestations. For each organ/system, major and minor clinical criteria are defined. In the absence of a family and genetic history, a diagnosis can be made if the patient has major criteria in at least two different organs/systems and the involvement of a third organ/system [5]. Simplification of diagnostic criteria is beneficial to low-income countries, allowing thus easy and early diagnosis in order to prevent complications. We could not perform genetic and family investigations for our patient. The diagnosis was based on dilation and dissection of the aorta, associated with many other systemic manifestations (deformity of the hindfoot, protrusion acetabulae, reduced upper to lower segment ratio, scoliosis, flat foot, mitral regurgitation, aneurysm and dissection of abdominal aorta). Besides the simplification of disease diagnosis, survival of patients with Marfan syndrome was extended by 30 years, due to therapeutic advances combining the proscription of violent sports, pharmacologic treatment with beta-blockers, ultrasound monitoring of aortic diameter and prophylactic surgery [3,12-14]. Indeed, ultrasound monitoring of aortic diameters allows early surgical procedures, in order to prevent aortic dissection which is the main cause of mortality [3].
The challenge is to define with accuracy at which stage of aortic dilation the surgery should be done and to describe the surgical procedure applied to the aortic valve.
According to the 2012 ESC recommendations for Marfan syndrome, aortic dilation of 50 mm is the threshold diameter indicated for surgery. However, the threshold of 45 mm remains valid with a Class IIa recommendation, in the presence of risk factors for dissection (expansion of aortic diameter > 2 mm per year, family history of dissection, desire for pregnancy, absence of treatment with beta-blocker, significant aortic or mitral regurgitation) [15].
Our patient underwent surgery using the Tirone David procedure [16]. The aim of this procedure is to spare the aortic valve when it appears healthy or slightly degenerated, in order to avoid the constraints of permanent anticoagulation. The procedure is technically fastidious and requires an experienced surgeon. The risk is the onset of aortic regurgitation which could require a new operation [17]. The use of this procedure in our case was therefore controversial, because of the presence of severe aortic regurgitation prior to surgery.
Bentall operation has become the procedure of choice nowadays [10]. In the study of Linggen Gao, et al. on prognostic factors after aortic surgery in Marfan patients, the Bentall procedure was more used than the Tirone David one (73.6% versus. 8%) [17]. However, because of the replacement of the native valve by a mechanical valve, Bentall’s operation requires lifelong anticoagulation and regular INR monitoring. The choice of Tirone David procedure for our patient was made by the surgeons of the philanthropic sponsor, probably because of his poor financial conditions. Indeed, he could not have been able to afford lifelong anticoagulation.
High systolic blood pressure and active smoking are factors of long-term poor postoperative prognosis [17,18]. Active smoking in our patient was an additional risk factor for his iterative aortic dissections, and remained also a factor of poor post-operative prognosis; he, therefore, needed very close monitoring.
The clinical feature of this patient was the occurrence of two dissections of different chronologies and topographies. He was at high risk of iterative surgery because of the first surgical procedure that was inappropriate for his clinical presentation, and because of persistent risk factors like cigarette smoking. In the context of poor conditions of medical practice, possibilities for re-intervention are doubtful; most patients with aortic dissection die early because of inadequate facilities. This was the case for our patient who could not afford medical follow-up because of a lack of health insurance. The life expectancy of patients with Marfan syndrome has increased from a mean age of 32 in 1972 to 41 in 1993, due to better disease comprehension, closer monitoring, active medical management and advances in cardiovascular surgical procedures [8,15].
Despite the known genetic patterns of Marfan syndrome, there is often a lack of standardized genetic counseling and screening, for family members of patients. Monitoring of aortic diameter, and surgical management of type A dissection are well codified by the current medical recommendations.
Marfan syndrome is a rare and unrecognized condition in our African context. We reported the case of a patient with two aortic dissections of different sites and chronologies. This happened because of inappropriate diagnosis and management of the pathology. Recent advances have improved patient survival, and scientific societies have given thresholds for aortic dilation, requiring prophylactic surgery. It is therefore worthy of monitoring steady aortic diameters on echography. The surgery is well codified and Bentall operation is the procedure of choice despite the need for lifelong anticoagulation. The long-term postoperative prognosis depends on many risk factors including active smoking. Our patient condition required multidisciplinary management and psychological support, not always available in our context. It raises the need to set up social groups and associations for supporting patients. Screening and care of family members should also be offered.
- Hecht F, Beals RK. "New" syndrome of congenital contractural arachnodactyly originally described by Marfan in 1896. Pediatrics. 1972 Apr;49(4):574-9. PMID: 4552107.
- Dietz HC, Cutting GR, Pyeritz RE, Maslen CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ, Curristin SM, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991 Jul 25;352(6333):337-9. doi: 10.1038/352337a0. PMID: 1852208.
- Dietz H, Francke U, Furthmayr H, Francomano C, De Paepe A, Devereux R, Ramirez F, Pyeritz R. The question of heterogeneity in Marfan syndrome. Nat Genet. 1995 Mar;9(3):228-31. doi: 10.1038/ng0395-228. PMID: 7773282.
- Faivre L, Collod-Beroud G, Adès L, Arbustini E, Child A, Callewaert BL, Loeys B, Binquet C, Gautier E, Mayer K, Arslan-Kirchner M, Grasso M, Beroud C, Hamroun D, Bonithon-Kopp C, Plauchu H, Robinson PN, De Backer J, Coucke P, Francke U, Bouchot O, Wolf JE, Stheneur C, Hanna N, Detaint D, De Paepe A, Boileau C, Jondeau G. The new Ghent criteria for Marfan syndrome: what do they change? Clin Genet. 2012 May;81(5):433-42. doi: 10.1111/j.1399-0004.2011.01703.x. Epub 2011 Jun 2. PMID: 21564093.
- Loeys BL, Dietz HC, Braverman AC, Callewaert BL, De Backer J, Devereux RB, Hilhorst-Hofstee Y, Jondeau G, Faivre L, Milewicz DM, Pyeritz RE, Sponseller PD, Wordsworth P, De Paepe AM. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010 Jul;47(7):476-85. doi: 10.1136/jmg.2009.072785. PMID: 20591885.
- Higurashi M, Oda M, Iijima K, Iijima S, Takeshita T, Watanabe N, Yoneyama K. Livebirth prevalence and follow-up of malformation syndromes in 27,472 newborns. Brain Dev. 1990;12(6):770-3. doi: 10.1016/s0387-7604(12)80004-0. PMID: 2092586.
- Grimes SJ, Acheson LS, Matthews AL, Wiesner GL. Clinical consult: Marfan syndrome. Prim Care. 2004 Sep;31(3):739-42, xii. doi: 10.1016/j.pop.2004.04.005. PMID: 15331256.
- Chan YC, Ting CW, Ho P, Poon JT, Cheung GC, Cheng SW. Ten-year epidemiological review of in-hospital patients with Marfan syndrome. Ann Vasc Surg. 2008 Sep;22(5):608-12. doi: 10.1016/j.avsg.2008.04.005. Epub 2008 Jun 17. PMID: 18562163.
- Nollen GJ, Groenink M, Tijssen JG, Van Der Wall EE, Mulder BJ. Aortic stiffness and diameter predict progressive aortic dilatation in patients with Marfan syndrome. Eur Heart J. 2004 Jul;25(13):1146-52. doi: 10.1016/j.ehj.2004.04.033. PMID: 15231373.
- Bentall H, De Bono A. A technique for complete replacement of the ascending aorta. Thorax. 1968 Jul;23(4):338-9. doi: 10.1136/thx.23.4.338. PMID: 5664694; PMCID: PMC471799.
- Januzzi JL, Isselbacher EM, Fattori R, Cooper JV, Smith DE, Fang J, Eagle KA, Mehta RH, Nienaber CA, Pape LA; International Registry of Aortic Dissection (IRAD). Characterizing the young patient with aortic dissection: results from the International Registry of Aortic Dissection (IRAD). J Am Coll Cardiol. 2004 Feb 18;43(4):665-9. doi: 10.1016/j.jacc.2003.08.054. PMID: 14975480.
- Grimes SJ, Acheson LS, Matthews AL, Wiesner GL. Clinical consult: Marfan syndrome. Prim Care. 2004 Sep;31(3):739-42, xii. doi: 10.1016/j.pop.2004.04.005. PMID: 15331256.
- Silverman DI, Burton KJ, Gray J, Bosner MS, Kouchoukos NT, Roman MJ, Boxer M, Devereux RB, Tsipouras P. Life expectancy in the Marfan syndrome. Am J Cardiol. 1995 Jan 15;75(2):157-60. doi: 10.1016/s0002-9149(00)80066-1. PMID: 7810492.
- Pyeritz RE. Marfan syndrome: 30 years of research equals 30 years of additional life expectancy. Heart. 2009 Mar;95(3):173-5. doi: 10.1136/hrt.2008.160515. Epub 2008 Nov 10. PMID: 19001001.
- Joint Task Force on the Management of Valvular Heart Disease of the European Society of Cardiology (ESC); European Association for Cardio-Thoracic Surgery (EACTS), Vahanian A, Alfieri O, Andreotti F, Antunes MJ, Barón-Esquivias G, Baumgartner H, Borger MA, Carrel TP, De Bonis M, Evangelista A, Falk V, Iung B, Lancellotti P, Pierard L, Price S, Schäfers HJ, Schuler G, Stepinska J, Swedberg K, Takkenberg J, Von Oppell UO, Windecker S, Zamorano JL, Zembala M. Guidelines on the management of valvular heart disease (version 2012). Eur Heart J. 2012 Oct;33(19):2451-96. doi: 10.1093/eurheartj/ehs109. Epub 2012 Aug 24. PMID: 22922415.
- David TE, Feindel CM. An aortic valve-sparing operation for patients with aortic incompetence and aneurysm of the ascending aorta. J Thorac Cardiovasc Surg. 1992 Apr;103(4):617-21; discussion 622. PMID: 1532219.
- Gao L, Zhou X, Zhang L, Wen D, Chang Q, Wu Y, Sun L, Hui R. Factors influencing prognosis in patients with marfan syndrome after aortic surgery. J Cardiothorac Vasc Anesth. 2011 Aug;25(4):625-31. doi: 10.1053/j.jvca.2010.11.019. Epub 2011 Jan 22. PMID: 21262573.
- Milewicz DM, Dietz HC, Miller DC. Treatment of aortic disease in patients with Marfan syndrome. Circulation. 2005 Mar 22;111(11):e150-7. doi: 10.1161/01.CIR.0000155243.70456.F4. PMID: 15781745.