
More Information
Submitted: 16 August 2019 | Approved: 02 September 2019 | Published: 03 September 2019
How to cite this article: Bejiqi R, Retkoceri R, Maloku A, Bejiqi R, Mustafa A, et al. How often is Klippel-Feil Syndrome associated with congential heart disease presentation of five cases and a review of the literature. J Cardiol Cardiovasc Med. 2019; 4: 110-116.
DOI: 10.29328/journal.jccm.1001050
ORCID ID: 0000-0002-5706-2363
Copyright License: © 2019 Bejiqi R, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Cervical vertebra fusion syndrome; Klippel-Feil syndrome; Short neck; Low hairline; Congenital heart disease

How often is Klippel-Feil Syndrome associated with congential heart disease presentation of five cases and a review of the literature
Ramush Bejiqi1, Ragip Retkoceri2, Arlinda Maloku3, Rinor Bejiqi4, Aferdita Mustafa3, Faruk Husremovic5 and Samir Bejic*6
1Visiting Professor, Health Science Centre, University of Texas, San Antonio, Texas, USA
2Associate Professor, University of Gjakova, Gjakova, Republic of Kosovo
3Division of Cardiology, Pediatric Clinic, University Clinical Centre of Kosovo, Prishtina, Republic of Kosovo
4Medical Faculty, University of Prishtina, Prishtina, Republic of Kosovo
5Cantonal Hospital, Zenica, Bosnia and Herzegovina
6Main Centre of Primary Medical Health Care, Sarajevo, Bosnia and Herzegovina
*Address for Correspondence: Samir Bejic, Main Centre of Primary Medical Health Care, Sarajevo, Bosnia and Herzegovina, Tel: +387 61 467 802; Email: [email protected]
Introduction: Klippel-Feil syndrome (KFS), is a bone disorder characterized by the abnormal joining (fusion) of two or more spinal bones in the neck (cervical vertebrae), which is present from birth. Three major features result from this abnormality: a short neck, a limited range of motion in the neck, and a low hairline at the back of the head. In some individuals, KFS can be associated with a variety of additional symptoms and physical abnormalities which contribute in the deterioration and complication of the condition of the child.
Aim of presentation: Here, we report five children from Kosovo with KFS associated with different heart abnormalities, clinical presentation, diagnosis, management, and outcomes of selected conditions in resources-limited settings.
Methods: Retrospectively we analysed medical reports of five children, diagnosed at different age with congenital disease and clinical and lab signs of Klippel-Feil syndrome.
Conclusion: Basing on our cases, all diagnosed in a small country as a Kosovo, we can conclude that KFS is not such a rare condition. In addition, such syndrome is not so rarely associated with different congenital heart disease. In four cases cardiac surgery was indicated and successfully was done abroad Kosovo in the lack of such services in Kosovo.
Klippel–Feil syndrome (KFS), known also with synonyms: cervical vertebra fusion syndrome, Klippel-Feil deformity, Klippel-Feil sequence disorder), is a rare disease, initially reported in 1884 by Maurice Klippel and André Feil from France, characterized by the congenital fusion of any two of the seven cervical vertebrae. Since first classification from Feil in three categories (I-III) other classification systems have been advocated to describe the anomalies, predict the potential problems, and guide treatment decisions. Patients with Klippel-Feil syndrome usually present the disease during childhood, but may present later in life. The challenge to the clinicians is to recognize the associated anomalies that can occur with Klippel-Feil syndrome and to perform the appropriate workup for diagnosis [1]. The clinical presentation of Klippel-Feil syndrome is varied because of the different associated syndromes and anomalies that can occur in patients with this syndrome. A complete history and careful physical examination may reveal some associated anomalies.
Klippel-Feil syndrome is a rare skeletal disorder primarily characterized by abnormal union or fusion of two or more bones of the spinal column (vertebrae) within the neck (cervical vertebrae). For the first time, it has been described in 1912, independently from Maurice Klippel and Andre Feil. Three major features result from this abnormality: a short neck, a limited range of motion in the neck, and a low hairline at the back of the head. In some individuals, KFS can be associated with a variety of additional symptoms and physical abnormalities but very rare with structural abnormalities of the heart - congenital heart defects [2].
Klippel-Feil syndrome (KFS) is a congenital anomaly characterized by a defect in the formation or segmentation of the cervical vertebrae, resulting in a fused appearance. The clinical triad consists of short neck, low posterior hairline, and limited neck movement, although less than 50% of patients demonstrate all 3 clinical features. However, despite the lack of accurate epidemiological data from Kosovo, KFS and variety are rare in children and adults on this region. KFS is usually non-malignant but causes significant morbidity, especially when is associated with anomalies in vital organs [3]. Discussion of unique aspects of etiology, diagnosis, and management in underserved regions of Kosovo may be important to raise awareness in health professionals, allows timely diagnosis, and improves management and prognosis. Here, we present four cases of patients with KFS and congenital heart disease, diagnosed at different age, surgical treatment and outcomes in Kosovo as a country with limited resources [4].
Case 1
A 2- days old male infant, weighing 3.5 kilograms, was noted to have bluish discoloration over the lips and periphery, with percutaneous oxygen saturation of 75%. The baby was born in Madrid from parents refugees from Kosovo during the war 1999. He required intubation following one apneic episode. Despite being ventilated on 100% oxygen, pulse oximetry could not be raised above 80%. Hence, a prostaglandin infusion was commenced. Physical examination showed a short neck, low hairline at the back of the head, and particularly restricted mobility of the upper spine. Cardiovascular and respiratory examination was normal. An electrocardiogram showed tight atrial and ventricular hypertrophy. Chest X-ray showed cardiomegaly. Transthoracic echo cardiography showed confluence of the all pulmonary veins above left atrium and draining together to the right atrium. The diagnosis of total anomalous venous return to the right atrium was confirmed. In the region of fosa ovale restrictive secundum atrial septal defect was presented with a predominantly right-to-left shunt. Suspicion for KFS was indication for cervical radiography and multiple developmental anomalies of the upper cervical spine, consistent with Klippel-Feil syndrome. Antero posterior (AP) as well as lateral neutral, flexion, extension radiographs of the cervical spine demonstrate abnormal development of the upper cervical spine described as follows: There is at least a partial fusion of C2 and C3, the ossified portion of the dens is noted to be inferiorly displaced relative to the anterior arch of C1, the posterior elements of C1, C2 and likely C3 are hypoplastic/absent, and there is a defect in the lamina of the superior-most vertebral body which contains formed posterior elements. The fused vertebral bodies are small and also have an abnormal anterior convex configuration. On the flexion and extension radiographs and atlantoaxial, instability is identified. The skull projects over most of the cervical spine on the AP views so evaluation for vertebral bodies is limited. The posterior skull is noted to have a “beaten copper” appearance. A cervical rib is present on the right. The paravertebral soft tissues have normal appearance. The nasopharynx, oropharynx, and laryngopharynx are patent and clear. Family history was normal for congenital heart disease, clefts, MR/DD, seizures, hearing loss, eye, Dx, diabetes, other birth defects, and pregnancy losses. Consanguinity is denied. Genetics examination confirmed new mutations in the GDF6. At the 7th day of birth, at the “Hospital Universitario La Paz” in Madrid surgical repair of the anomalous pulmonary venous return was done successfully and in the next day, in stable condition, was extubated. At the 15th day of life he was discharged on the only Captopril, 0.5mg/kg three times daily. Control echocardiography showed normal pulmonary venous return without obstruction and normal heart function (Figure 1).
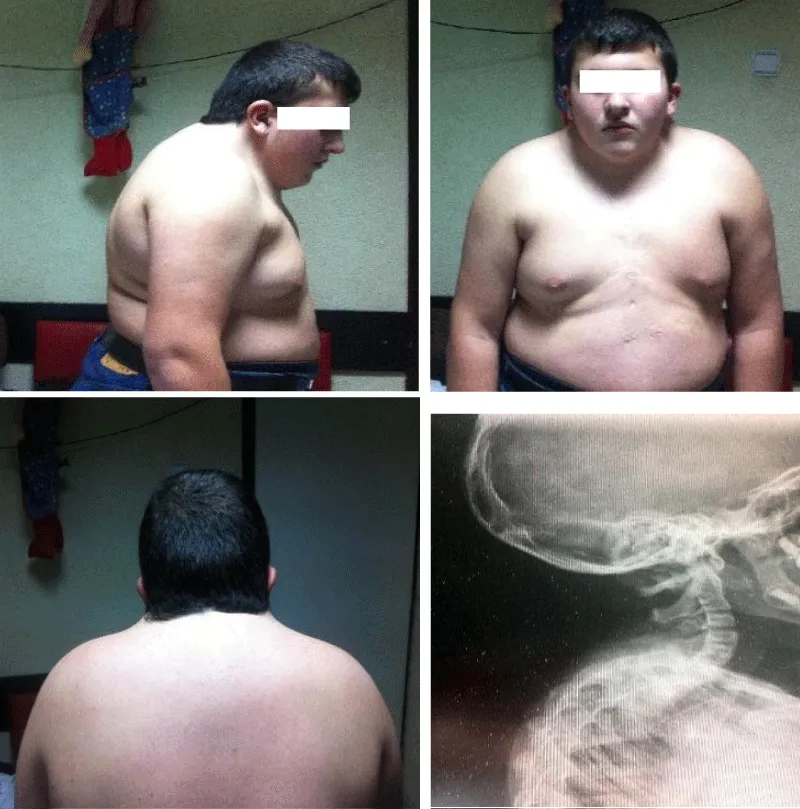
Figure 1: Case 1: A 15-years-old boy diagnosed at delivery with Klippel-Feil syndrome. Note the short neck, low hairline and symmetric shoulders. (Scar on the thorax after surgical repair of total anomalous pulmonary venous return).
Case 2
A 28-month-old girl, as the fourth child, from normal pregnancy and absolutely health parents, weighing 16.3 kg, during the routine pediatric examination systolic murmur was noted and for cardiological examination at tertiary level was referred. The child’s growth and development was completely normal. There was no sweating or fatigue during feeding or normal activities. Complete clinical and cardiological examination was obtained. Objective examination of other children is in normal limits. During routine examination a short neck, low hairline at the back of the head and restricted mobility of the upper spine was noted. Anteroposterior (AP) and lateral in extension radiographs of the cervical spine demonstrate abnormal development of the upper cervical spine as a partial fusion of C2 and C3 and hypoplastic C1 spine. The fused vertebral bodies manifested an abnormal anterior convex configuration. Arterial blood gas was within normal limits. Clinical examination demonstrated: a quite precordium, normal first heart sound, short midsystolic murmur 2-3/6 degree on the apex and left sternal border, and single second heart sound. Electrocardiogram showed: normal sinus rhythm, left axis deviation, and incomplete bundle branch block. A chest radiogram revealed a normal cardiac silhouette. The pulmonary vasculature was normal in appearance, and there were no infiltrates seen. Cross-sectional echocardiography demonstrated: normal systemic and pulmonary vein connection, big hole, diameter 12mm, in the middle part of interatrial septum with non-restrictive left to right flow. There was normal atrio-ventricular and ventriculo-arterial connection. By continuous Doppler waves and color Doppler trivial tricuspid regurgitation and hyper dynamic flow through the pulmonary, artery was noted. Family history for consanguinity and congenital heart disease is denied. At age of 18 month, in the absence of cardiac services in Kosovo, child was sent in the Santa Rosa Children’s Hospital (USA) and surgical correction of atrial septal defect has been successfully done. Also genetics examination confirmed new mutations in the GDF6 and GDF3 genes (Figures 2,3).
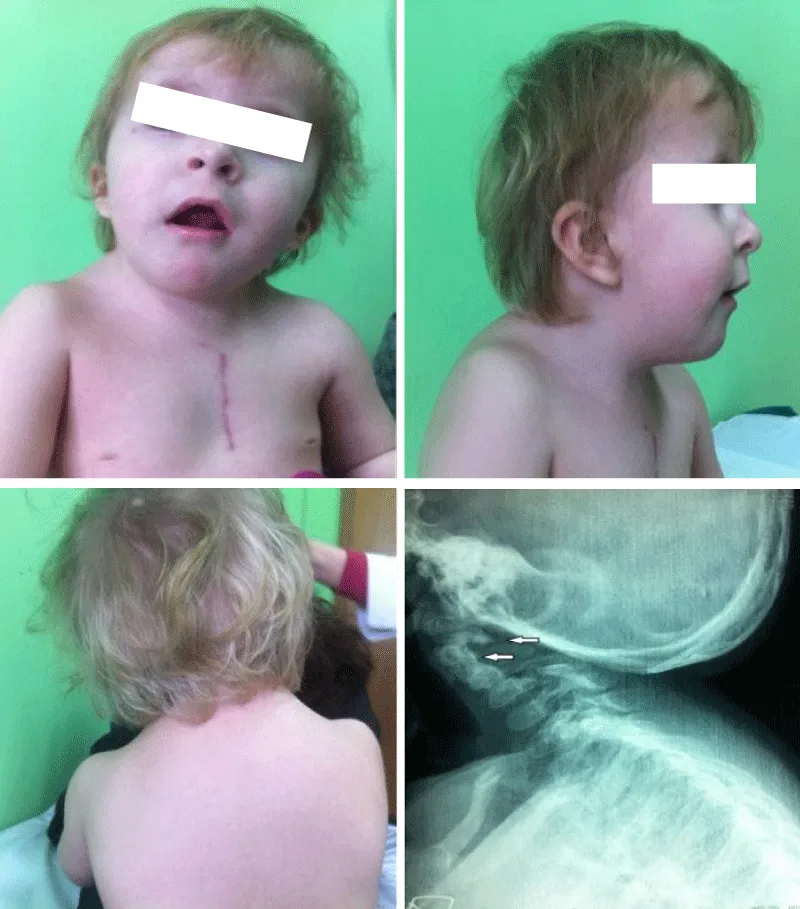
Figure 2: Case 2: Patient with Klippel-Feil syndrome and anomaly of the occipito-cervical junction. The images show an elevated left shoulder due to a Sprengel anomaly, a short, webbed neck, and a low hairline. (Scar on the thorax after surgical repair of nonrestrictive atrial septal defect). X-ray shows occipito-cervical junction.
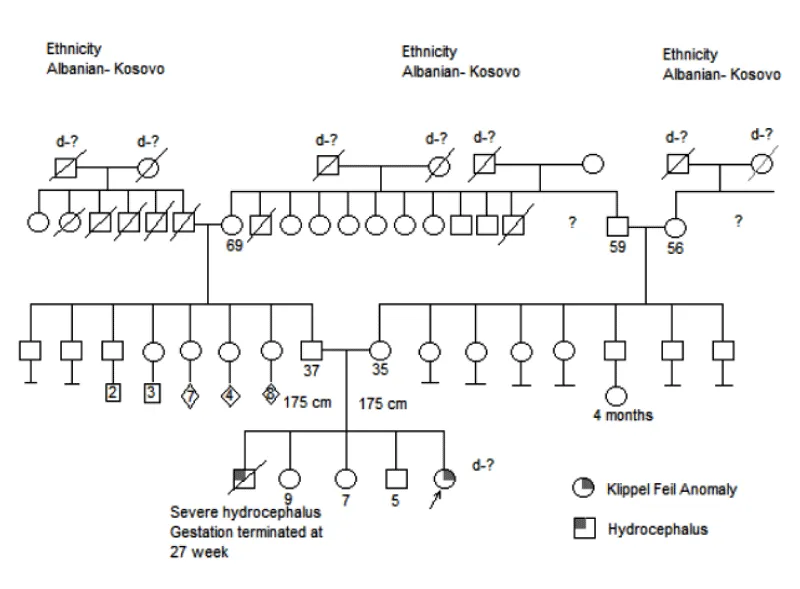
Figure 3: Family tree of Case 2.
Case 3
A 2-day-old male infant, weighing 2890 g., from the first and normal pregnancy, from both healthy parents, born in regional hospital, at the second day of life is presented with respiratory distress. Despite being ventilated on 100% oxygen, pulse oximetry could not be raised above 85%. Baby was transferred at tertiary level and a complete history and careful physical examination was obtained. Objectively a short, webbed neck, decreased range of motion in the cervical spine and a low hairline was noted and suspicion for KFS awakened. Anteroposterior (AP) and lateral radiographs of the cervical spine demonstrate abnormal development of vertebra C1, C2 and C3 with massive fusion of the cervical spine. Echocardiography revealed situs solitus, levocardia, atrioventricular concordance and transposition of the great arteries with patent ductus arteriosus. At the third day of life balloon atrioseptostomy was performed and 7-8mm communication between atria was achieved. In the lack of cardio surgery services in Kosovo and financial impossibility of family to cover surgery abroad Kosovo, baby dies after 2 months as a consequence of congestive heart failure and respiratory infection (Figure 4).
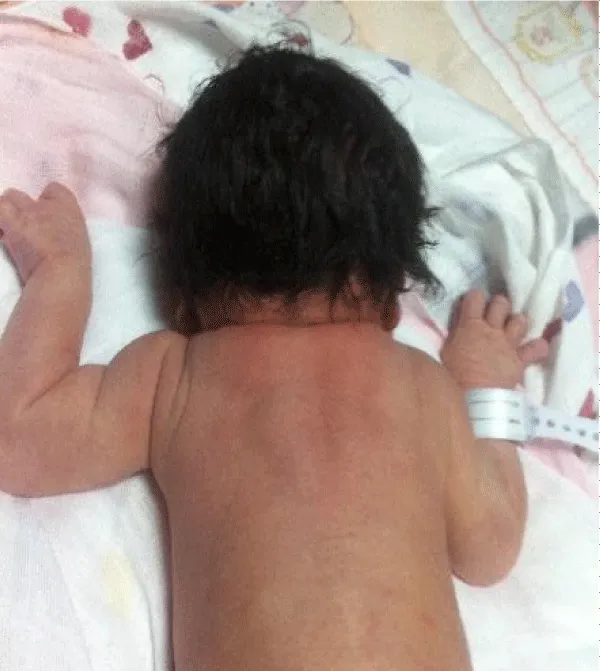
Figure 4: Case 3. A 2-days old neonate with Klippel-Feil syndrome. The image shows an elevated right shoulder due to a Sprengel anomaly, a short, webbed neck, and a low hairline.
Case 4
An asymptomatic girl, aged 12 years, born to non-consanguineous parents and from normal pregnancy, so far with normal development, was referred for assessment of a recently discovered systolic cardiac murmur, grade at 2 from 6, and a best heard along the left parasternal border. During the clinical examination a very short neck, reduced bilateral neck movements, and a low hairline at the back of the head was noted. Radiographic examination showed fusion of C1 and C2 and C5-7 vertebrae. Transthoracic echocardiographic examination, using Acuson Sequoia 256 machine, showed usual arrangement of the atrial appendages and thoraco-abdominal organs, with concordant atrioventricular and ventriculo-arterial connections. There was a slight enlargement of the right heart side. In the parasternal short-axis view thick pulmonary valve cusp with restricted systolic motion was noted. The main pulmonary artery was dilated. By Doppler and color Doppler turbulent flow through the pulmonary, artery was noted with max pressure of 68mm of mercury (Figure 5).
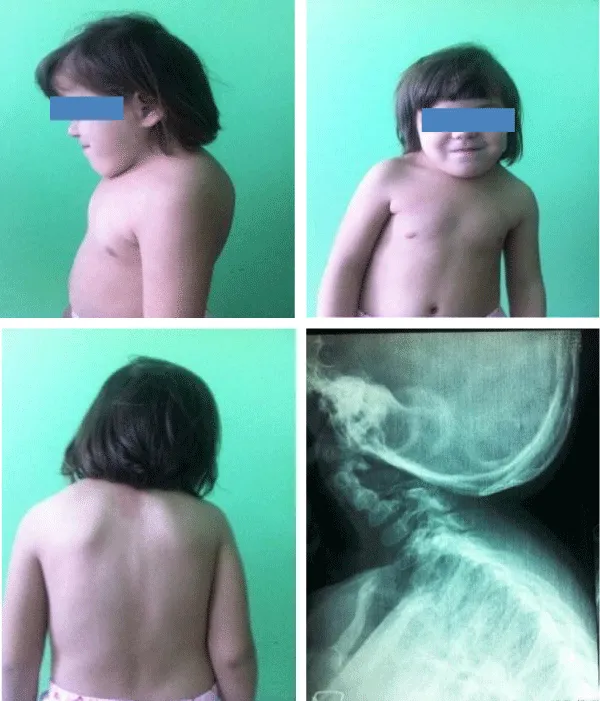
Figure 5: Case 4. A 12-years-old girl with Klippel-Feil syndrome and pulmonary stenosis.Note elevated left shuoldres and thoarx deformity.
Case 5
A 5-day-old male infant, with initials GS, from the fourth and normal controlled pregnancy, weighing 3520g, from both healthy parents, was transferred from regional hospital at our centre as a reason of central cyanosis and important heart murmur. Delivery was normal, with normal Apgar score and despite being ventilated on 100% oxygen, pulse oximetry could not be raised above 88%. Objectively a short, webbed neck and a low hairline and suspicion were for Klippel-Feil syndrome. A complete history and careful physical examination was obtained including X-ray chest and transthoracic echocardiography examination. Echocardiography presented severe form of tetralogy of Fallot. Anteroposterior (AP) and lateral radiographs of the cervical spine was done which presented abnormal development of C1, C2 and C3 vertebra with massive fusion of the cervical spine. At age of four month for surgical intervention in Linz, Austria was sent, where complete correction of tetarlogy of Fallot was done. Genetic testing confirmed new mutations in the GDF6 gene. At age of 7 years as a consequence of huge pulmonary insufficiency Melody valve was implanted at the same centre. Actual medical status is well, within normal school and other physical activities (Figure 6).
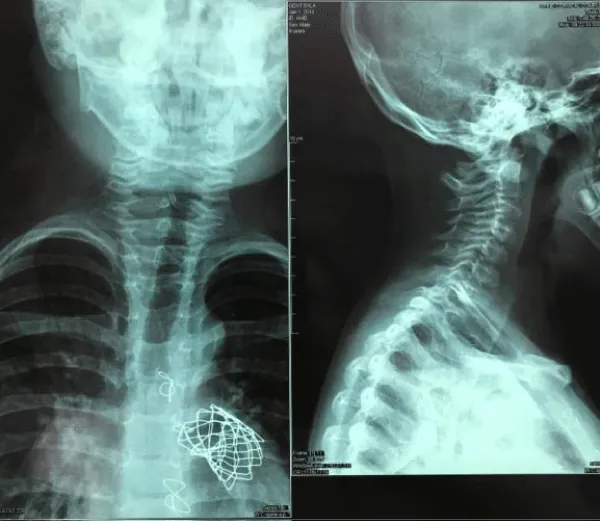
Figure 6: Case 5. X-ray images shows occipito-cervical junction.Note Melody valve implanted as a consequence of pulmonary insufficiency after complete TOF repair.
Incidence
The actual occurrence for the KFS syndrome is unknown; it is estimated to occur 1 in 40,000 to 42,000 new-borns worldwide. In addition, females seem to be affected slightly more often than males. Mutations in the GDF6 and GDF3 genes can cause Klippel-Feil syndrome. These genes provide instructions for making proteins that belong to the bone morphogenetic protein family, which is involved in regulating the growth and maturation (differentiation) of bone and cartilage. Additional forms of KFS include autosomal recessive KFS2 (214300), caused by mutation in the MEOX1 gene (600147) on chromosome 17q21, and autosomal dominant KFS3 (613702), caused by mutation in the GDF3 gene (606522) on chromosome 12p13. Sometimes this condition is inherited in an autosomal recessive pattern, which means both copies of a gene in each cell have mutations. However, in these cases, the gene involved is unknown. The parents of an individual with an autosomal recessive condition each carry one copy of the mutated gene, but they typically do not show signs and symptoms of the condition [5,6].
Etiology
The etiology of Klippel-Feil syndrome and its associated conditions is unknown. The syndrome can be presented with a variety of other clinical syndromes, including fetal alcohol syndrome, Goldenhar syndrome, anomalies of the extremities etc. The occurrence of KFS in siblings and in consanguineous families suggests autosomal recessive inheritance of a form of the disorder. A close evaluation of the immediate family is indicated, because autosomal dominant inheritance with variable expression in affected individuals has been noted, although this is presumably rare. There is a strong association with congenital abnormalities of the genitourinary tract (30-40%), including double collecting systems, renal aplasia and horseshoe kidney [7].
Data of our patients are presented on the table 1.
| Table 1: Demographic data of presented patients CHD-Congenital Heart Defect, N of CVF-Number of Cervical Vertebra Fusion | ||||||
| Number | Age at Diagnosis | Gender | CHD | N of CVF | Actual Age | Outcome |
| 1 | 2 Days | Male | TAPVR | 3 (C1,C2,C3) | 19 years | Alive |
| 2 | 28 Months | Female | ASD II | 3 (C1,C2,C3) | 14 years | Alive |
| 3 | 2 Days | Male | D-TGA | 3 (C1,C2,C3) | 0 | Died |
| 4 | 12 Years | Female | St AP | 5 (C1-C2,C5-C7) | 15 years | Alive |
| 5 | 2 Months | Male | TOF | 2 (C1,C2) | 13 years | Alive |
| Table 2: Review of literature | |||
| Lead Author | Year | No of Patients | Siblings |
| Lubs [18] | 1963 | 2 from 11 | parents consanguineous |
| Juberg and Gershanik [19] | 1976 | 1 | parents consanguineous |
| Chemke [20] | 1980 | 1 | parents no consanguineous |
| Da-Silva [21] | 1982 | 4 siblings (7f/5m) | parents consanguineous |
| Fragoso [22] | 1982 | 1 | parents no consanguineous |
| Clarke [23] | 1998 | Family | autosomal recessive inheritance |
| Bejiqi [24] | 2013 | 1 | parents no consanguineous |
| Erol [25] | 2004 | 1 | parents consanguineous |
| Ohashi[26] | 1992 | 1 | de novo balanced translocation |
| Bejiqi [27] | 2015 | 4 | parents no consanguineous |
These cases reported show several aspects of KFS in different ages of young Kosovars. They highlight common clinical presentation and etiology, as well as the challenges for the diagnosis and management of these clinical entities resources- poor settings. KFS has diverse clinical presentation and etiology. The presenting symptoms and signs depend on the age at presentation (at birth-congenital type), on the primary disease (abnormality of the neck and restricted movement of the head and neck) as well as the associated anomalies, especially heart anomalies. Associated anomalies also may include abnormal curvature of the spine (scoliosis) and/or vertebral instability, spina bifida occulta, raised scapula (Sprengel’s deformity), absent rib(s) and other rib defects including cervical ribs, other skeletal abnormalities including skeletal malformations of the ear, nose, mouth and larynx including hearing impairment and cleft palate, malformations of the head and facial (craniofacial) area; anomalies of the urinary tract and/or kidney including absent or horse-shoe kidney; or structural abnormalities of the heart (congenital heart defects), mirror movements, webbing of the digits and digital hypoplasia. In addition, in some cases, neurological complications may result due to associated spinal cord injury. The challenge to the clinician is to recognize the associated anomalies that can occur with Klippel-Feil syndrome and to perform the appropriate workup for diagnosis [8-10].
Since the first classification from Feil in three categories (I-III) other classification systems have been advocated to describe the anomalies, predict the potential problems, and guide treatment decisions. Patients with Klippel-Feil syndrome usually are presented with the disease during childhood, but may present later in life [11,12]. Two overlapping classification systems exist for KFS. The original classification of KFS subtypes (I, II and III) described by Maurice Klippel and Andre Feil and one more recent updated classification of KFS Classes (KF1-4) as described by Clarke and colleagues.
It is important to note that affected individuals will not have all of the symptoms discussed below.
The original classification differentiate individuals based on the degree of cervical fusion; KFS type I is characterized by extensive fusion of vertebrae of the neck (cervical vertebrae) and the upper back (i.e., upper thoracic vertebrae); type II is characterized by fusion at one or two cervical or thoracic vertebrae; type III is characterized by fusion of vertebrae of the neck as well as vertebrae of the upper or lower back (i.e., lower thoracic or lumbar vertebrae). In contrast, the classification described by Clarke at all differentiate individuals based on patterns of inheritance, common associated anomalies and the axial level of the most anterior fusion. KF1 is the only class presenting with C1-2 fusion, the very short neck and recessive inheritance of type I, II or III fusion patterns; KF2 is autosomal dominant with the most anterior fusion at C2-3 in association with type I, II or III fusion patterns; KF3 is recessive or has reduced penetrance of isolated fusions between any of the cervical vertebrae except C1-2; KF4 includes cases of Wildervank and Duane syndrome [13-15]. The specific symptoms associated with KFS vary greatly from one person to another. Historically, KFS was associated with a classic triad of symptoms, specifically abnormally short neck, restricted movement of the head and neck, and a low hairline at the back of the head (posterior hairline). However, researchers have determined that these symptoms may define one class of KFS, which accounts for less than 50 percent of affected individuals [16]. Approximately 30 percent of affected individuals have additional skeletal abnormalities, such as fusion of certain ribs or other rib defects; abnormal sideways curvature of the spine (scoliosis); or a condition known as Sprengel’s deformity. Also, in some individuals with KFS, a portion of the spinal cord may be exposed due to incomplete closure of certain vertebrae (spina bifida occulta) [17]. In our cases we didn’t found additional skeletal abnormalities or spina bifida.
Approximately 25 to 50 percent of individuals with KFS also have hearing impairment. Such hearing loss may result from impaired transmission of sound from the outer or middle ear to the inner ear (conductive hearing loss); failed transmission of sound impulses from the inner ear to the brain (sensorineural hearing loss); or both (mixed hearing loss). Various eye (ocular) abnormalities may also be associated with KFS, such as deviation of one eye toward the other (cross-eye or convergent strabismus); involuntary, rapid eye movements (nystagmus), or absence or defects of ocular tissue (colobomas) [18,19]. In three of our cases we found eyes deviation and strabismus while in one of them ocular coloboma was noted. In addition, some affected individuals may have other abnormalities of the head and facial (craniofacial) area including facial asymmetry, in which one side of the face appears dissimilar from other side, with one eye higher than the other. There may also be abnormal twisting of the neck (torticollis), causing the head to be rotated into an abnormal position. According to some reports, approximately 17 percent of individuals with KFS also have incomplete closure of the roof of the mouth (cleft palate) [20]. In our cohort group we found in two cases facial asymmetry (Figure 4). KFS may sometimes be associated with additional physical abnormalities. These may include structural malformations of the heart (congenital heart defects), particularly ventricular septal defects (VSDs). VSDs are characterized by the presence of an abnormal opening in the fibrous partition (septum) that separates the two lower chambers of the heart. Some individuals may also have kidney (renal) defects, such as underdevelopment (hypoplasia) or absence (agenesis) of one or both kidneys; abnormal renal rotation or placement (ectopia); or swelling of the kidneys with urine (hydronephrosis) due to blockage or narrowing of the tubes (ureters) that carry urine to the bladder [21].
Some individuals with the disorder may also develop neurological complications due to associated spinal cord injury. Associated neurological complications tend to develop between the second and third decades of life and may occur spontaneously or following minor trauma. Such complications may include pain; abnormal sensations (paresthesia), such as tingling, prickling, or burning; or involuntary muscle movements accompanying certain voluntary actions (synkinesia or mirror movements). Some individuals may develop increased reflex reactions (hyperreflexia); weakness or paralysis of one side of the body (hemiplegia) or of the legs and the lower part of the body (paraplegia); or impairment of certain nerves that emerge from the brain (cranial nerve palsies) [22,23]. In our presented group we didn’t find any neurological complications.
The disorder can be present at birth (congenital), but mild cases may go undiagnosed until later during life when symptoms worsen or first become apparent. For the first time, it has been described in 1912, independently from Maurice Klippel and Andre Feil. Three major features result from this abnormality: a short neck, a limited range of motion in the neck, and a low hairline at the back of the head [24]. In all our cases heart problems have been diagnosed at early neonatal period and also in two cases clinical symptoms of KFS have been recognised. Clinical presentation is varied because of the different associated syndromes and anomalies that can occur in patients with Klippel-Feil syndrome. A complete history and careful physical examination may reveal some associated anomalies. Klippel-Feil syndrome involves the congenital fusion (failure of segmentation) of one or more cervical motion segments, and most patients have associated congenital anomalies of the cervical spine or other organs and systems. These anomalies may occur at the cranio cervical junction (occipit-C2), the sub axial spine (below C2), or both. Our cases have multiple developmental anomalies of the upper cervical spine consistent with Klippel-Feil syndrome. The posterior skull has a “beaten copper” appearance which may represent a normal variant or less likely reflective of gyral impression from increased intracranial pressure because it is only seen posteriorly [18,25]. Associated anomalies occur in the auditory system, neural axis, cardiovascular system, and the musculoskeletal system. Cardiovascular anomalies, mainly septal defects, were found in 7 patients in Hensinger’s series, with 4 of these individuals requiring corrective surgery [26]. In our presentation all cases have a different CHD including those which from delivery threatening children’s life (first case with total anomalous pulmonary venous return). People with Klippel-Feil syndrome may have other features in addition to their spine abnormalities. Some people with this condition have hearing difficulties, genitourinary abnormalities such as malformed kidneys, a type of birth defect that occurs during the development of the brain and spinal cord (neural tube defect), an opening in the roof of the mouth (cleft palate), or heart abnormalities [27]. Consultations from different specialties in Kosovo and in both 1st and 2nd case during the surgical intervention abroad, other anomalies have been eliminated. Careful examinations of specialist exclude anomalies in other organs and systems. Radiographs and MRI of the thoracic and lumbosacral spine are obtained and other anomalies have been excluded.
Affected individuals may have underdeveloped shoulder blades that sit abnormally high on the back, a condition called Sprengel deformity [28]. In three of our cases we have found underdeveloped shoulder where two of them have an elevated left shoulder and the other one has the right. Lateral flexion-extension radiographs of the cervical spine should be performed on all patients to determine the motion of each open interspace. Clinically, flexion-extension is often maintained if single functioning open interspaces are maintained. Those with hyper mobility of the upper cervical segment are at risk of developing neurologic impairment. Affected individuals with hyper mobility of the lower cervical segment are at increased risk for degenerative disk diseases and should be treated symptomatically [29,30].
Limitation of study
Based on the fact that in all presented children diagnosis has been made in Kosovo, in the lack of accurate genetic laboratory and Magnetic resonance imaging machine, in some of them genetic examination and MRI examination has not been done.
In our study all cases were affected from different type of congenital heart disease, starting from easy forms (atrial septal defect and pulmonary stenosis), to life suffering from delivery (Tetralogy of Fallot and transposition of the great arteries). To the author’s best knowledge our study is the first publication which suggests the relationship between Klippel-Feil syndrome and different type of CHD, where prognosis is strongly connected with cardiac surgery in a country as a Kosovo where this service is absent.
- Samartzis D, Lubicky JP, Herman J. Symptomatic cervical disc herniation in a pediatric Klippel- Feil patient: the risk of neural injury associated with extensive congenitally fused vertebrae and a hypermobile segment. Spine. 2006; 31: E335-338. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/16688024
- Samartzis DD, Herman J, Lubicky JP, Shen FH. Classification of congenitally fused cervical patterns in Klippel-Feil patients: epidemiology and role in the development of cervical spine-related symptoms. Spine. 2006; 31: E798-80. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17023841
- Klimo P Jr, Rao G, Brockmeyer D. Congenital anomalies of the cervical spine. Neurosurg Clin N Am. 2007; 18: 463-478. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17678749
- Samartzis D, Kalluri P, Herman J, Lubicky JP, Shen FH. The extent of fusion within the congenital Klippel-Feil segment. Spine (Phila Pa 1976). 2008; 33: 1637-1642. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18594455
- Samartzis D, Lubicky JP, Herman J, Shen FH. Faces of Spine Care: From the Clinic and Imaging Suite. Klippel-Feil syndrome and associated abnormalities: the necessity for a multidisciplinary approach in patient management. Spine J. 2007; 7: 135-137. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17269206
- Tracy MR, Dormans JP, Kusumi K. Klippel-Feil syndrome: clinical features and current understanding of etiology. Clin Orthop Relat Res. 2004; 424: 183-190. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15241163
- Neidengard L, Carter TE, Smith DW. Klippel-Feil malformation complex in fetal alcohol syndrome. Am J Dis Child. 1978; 132: 929-930. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/685915
- Tassabehj M, Fang ZM, Hilton EN, Zhao Z. Mutations in GDF6 are associated with vertebral segmentation defects in Klippel-Feil syndrome. Hum Mutat. 2008; 29: 1017-1027. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18425797
- Sullivan JA. Klippel-Feil syndrome Medscape. 2016.
- Borujeni MJS, Purzaki M, Bakhtiari A, Sazegar G. A Rare Anatomical Variation of the Cervical Vertebrae Characterized by the Abnormal Fusion between C4 and C5. Global Journal of Medicine Researches and Studies. 2014; 1: 57-60.
- Stadnicki G, Rassumowski D. The association of cleft palate with the Klippel-Feil syndrome. Oral Surg Oral Med Oral Pathol. 1972; 33: 335-340. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/4501166
- Shankar VV, Kulkarni RR. Block vertebrae: Fusion of axis with the third cervical vertebrae-a case report. Int J Anatomical Variations. 2011; 4: 15-18.
- Krakov D. Spinal abnormalities and Klippel-Feil syndrome; Obstetric Imaging: Fetal Diagnosis and Care (second edition). 2018; 295-297.
- Samartzis D, Herman J, Lubicky JP, Shen FH, Jonathan JM, et al. Sprengel's deformity in Klippel-Feil syndrome. Spine. 2007; 32: E512-516. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC5822809/
- Drvaric DM, Ruderman RJ, Conrad RW, Grossman H, Webster GD, et al. Congenital scoliosis and urinary tract abnormalities: are intravenous pyelograms necessary? J Pediatr Orthop. 1987; 7: 441-443. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/3301897
- Auerbach JD, Hosalkar HS, Kusuma SK, Wills BP, Dormans JP, et al. Spinal cord dimensions in children with Klippel-Feil syndrome: a controlled, blinded radiographic analysis with implications for neurologic outcomes. Spine. 2008; 33: 1366-1371. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/18496350
- Yi S, Kim SH, Shin HC, Kim KN, Yoon DH. Cervical arthroplasty in a patient with Klippel-Feil syndrome. Acta Neurochir (Wien). 2007; 149: 805-809. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/17616843
- Baba H, Maezawa Y, Furusawa N, Chen Q, Imura S, et al. The cervical spine in the Klippel-Feil syndrome. A report of 57 cases. Int Orthop. 1995; 19: 204-208. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/8557413
- Thompson, E., Haan, E., Sheffield, L. Autosomal dominant Klippel-Feil anomaly with cleft palate. Clin Dysmorph. 1998; 7: 11-15. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9546824
- Lubs HA, Gunderson CH, Greenspan RH. Genetic re-evaluation of fused cervical vertebrae (Klippel-Feil anomaly). Clin. Res. 1963; 11: 179.
- Juberg RC, Gershanik JJ. Cervical vertebral fusion (Klippel-Feil) syndrome with consanguineous parents. J Med Genet. 1976; 13: 246-249. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/933127
- Chemke J, Nisani R, Fischel RE. Absent ulna in the Klippel-Feil syndrome: an unusual associated malformation. Clin Genet. 1980; 17: 167-170. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7363502
- Elias Oliveira DS. Autosomal recessive Klippel-Feil syndrome. J Med Genet. 1982; 19: 130-134. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1048843/
- Fragoso R, Cid-Garcia A, Hernandez A, Nazara Z, Cantu JM. Frontonasal dysplasia in the Klippel-Feil syndrome: a new associated malformation. Clin Genet. 1982; 22: 270-273. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/7151311
- Clarke RA, Catalan G, Diwan AD, Kearsley JH. Heterogeneity in Klippel-Feil syndrome: a new classification. Pediatr Radiol. 1998; 28: 967-974. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/9880643
- Bejiqi R, Retkoceri R, Bejiqi H, Zeka N, Maloku A, et al. Klippel - Feil Syndrome Associated with Atrial Septal Defect. Med Arh. 2013; 67: 141-142. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/24341064
- Erol M, Caksen H, Tan O, Atik B, Unal O, et al. Report of a girl with Klippel-Feil syndrome and Poland anomaly. Genet Counsel. 2004; 15: 469-472. PubMed: https://www.ncbi.nlm.nih.gov/pubmed/15658624
- McKusick VA. Online Mendelian Inheritance in Man (OMIM). Baltimore. MD: The Johns Hopkins University. 2014.
- Bejiqi R, Retkoceri R, Bejiqi H, Zeka N, Klippel-Feil Syndrome Associated with Congential Heart Disease - Presentation of Cases and a Review of the Curent Literature, OAMJMS. 2015; 129-134. PubMed: https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4877771/
- Angeli E, Wagner J, Lawrick E, Moore K, Anderson M, et al. General format title. 2010.