
More Information
Submitted: December 02, 2024 | Approved: December 16, 2024 | Published: December 17, 2024
How to cite this article: Prem AS, Shahanas PS, Sreekumar P, Ramaswamy NV. An Adult Case of Beta Thalassemia with Right Ventricular Outflow Tract Tachycardia: A Case Report J Cardiol Cardiovasc Med. 2024; 9(3): 177-179. Available from: https://dx.doi.org/10.29328/journal.jccm.1001201
DOI: 10.29328/journal.jccm.1001201
Copyright License: © 2024 Prem AS, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: β-thalassemia major; Iron overload; Right ventricular outflow tract tachycardia; Ventricular Tachycardia

An Adult Case of Beta Thalassemia with Right Ventricular Outflow Tract Tachycardia: A Case Report
Prem AS, Shahanas PS, Praveen Sreekumar* and Ramaswamy NV
Department of Cardiology and Hematology, Aster Medcity, Kochi, India
*Address for Correspondence: Dr. Praveen Sreekumar, Department of Cardiology and Hematology, Aster Medcity, Kochi, India, Email: [email protected]
Beta thalassemia major is a genetic disorder requiring recurrent blood transfusion leading to iron overload in endocrine glands and major organs like the heart. Iron overload in the heart may lead to many conduction abnormalities. This is a case report of a 20-year-old female who was on recurrent blood transfusion. She was on chelation therapy for iron overload. She developed Right Ventricular Outflow Tract Tachycardia (RVOT) which could not be managed with chemical or electrical cardioversion. Her condition was successfully managed with an Automatic Implantable Cardioverter Defibrillator (AICD) implantation and no ventricular tachycardia was observed even after four years of follow-up.
β-thalassemia is the most common genetic disorder, resulting in ineffective erythropoiesis leading to chronic hemolytic anemia [1]. The condition is diagnosed between 6 months to 2 years. Patients with β-thalassemia major often have severe anemia [2]. Reduced hemoglobin levels result in bone marrow expansion to compensate for the loss of red blood cells leading to bone abnormalities, spleen enlargement, and growth retardation [2]. Hence, red blood cell transfusion is necessary to maintain a hemoglobin level of 9.5 - 10.5 mg/dl which is required for the normal growth and development of the child until 10 - 11 years of age. Anemia management with blood products may result in iron overload in various organs causing endocrinopathies and features of secondary hemochromatosis including disruption of endocrine glands, dark metallic skin pigmentation, cirrhosis, cardiac arrhythmia, and myopathy. All these contribute to 71% of deaths in patients with thalassemia major [3]. After 10-11 years of age, the risk for posttransfusion iron overload increases resulting in severe cardiac, liver, and endocrine gland complications depending upon their compliance with chelation therapy [4]. The iron burden in the body increases by 2 g to 5 g per year compared to 0.0015g per year in healthy individuals [5]. Cardiac disease due to iron overload is the primary cause of mortality and morbidity in individuals with β-thalassemia. Here we report a case with cardiac hemosiderosis in a 20-year-old female who presented to the cardiac outpatient clinic with recurrent breathlessness.
A 20-year-old female patient from Malabar region in South India, presented with complaints of recurrent breathlessness and palpitations. She had a case of transfusion-dependent anemia and was diagnosed as β-thalassemia major at 18 months of age (Figure 1a,b). She underwent splenectomy at 11 years of age. At the time of presentation, she required transfusions every 20 to 35 days. She gave a history of iron overload features and was amenorrheic since 2014 due to secondary ovarian failure. She had a history of ventricular tachycardia and was on amiodarone, lasilactone, pentids, and aspirin. She had poor compliance to iron chelators and she was allergic to most chelators and sugar-coated pills. She was started on oral chelation therapy with Deferasirox1500 mg for iron overload as it was a cardio-protective agent for one year. Examination showed ECOG PS scale 1. Her vitals were stable. She had pallor and skin pigmentation. Abdominal examination showed an enlarged liver, tender. A splenectomy scar was present. There was joint tenderness and bone pains. Her complete blood count showed a low hemoglobin level of 7 mg/dl, and her ferritin level was 5800 ng/ml.
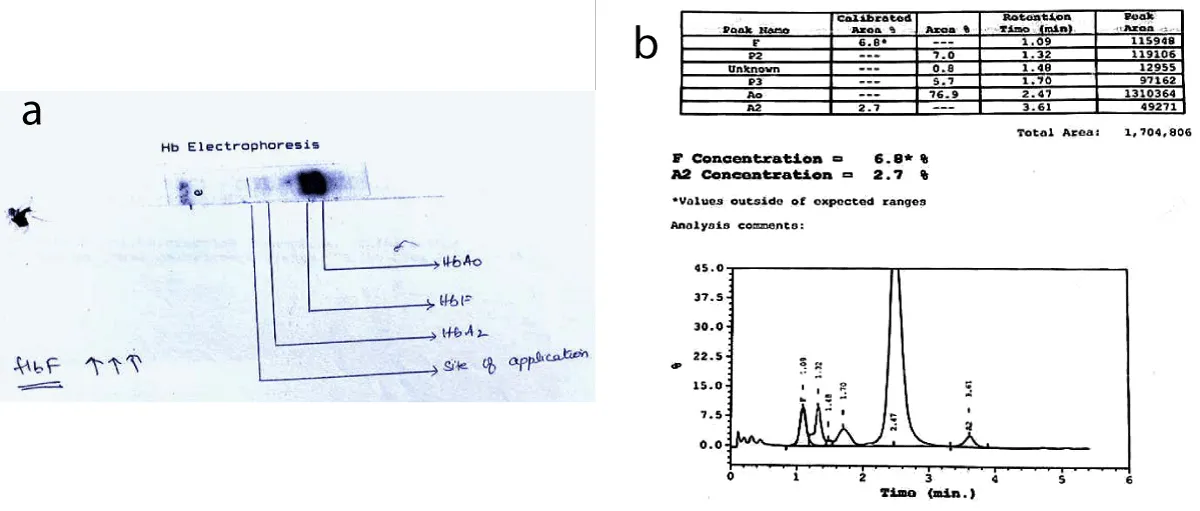
Figure 1: a: The gel electrophoresis pattern for Hemoglobin F(HbF) typically shows a distinct band or peak that corresponds to the HbF molecule. b: Hemoglobin electrophoresis shows significant elevation of hb A. x axis-time; y axis; Percentage of Total hemoglobin.
She was started on dual oral iron chelators as she was not keen on injectable. She was not able to tolerate deferiprone due to arthralgia and was started on intravenous deferoxamine. Her other systemic examinations were normal except for elevated jugular venous pressure, mild pedal edema, and irregular pulse rate with muffled heart sounds. Her echocardiography showed mildly dilated right atrium and right ventricle, severe biventricular dysfunction (LVEF 20%), moderate trivial regurgitation, and mild pulmonary artery hypertension. Her MRI heart and abdomen was suggestive of light iron overload in the liver and myocardium. Her 24-hour HOLTER monitor showed Right Ventricular Outflow Tract (RVOT) Ventricular Tachycardia (VT). She was started on amiodarone infusion. Even upon starting on chemical cardioversion, her heart rhythm was not under control and had to be electrically cardioverted. She also had chest pain radiating to her left arm and was diagnosed with first-degree Atrioventricular (AV) block and RVOT VT. Given recurrent episodes of ventricular tachycardia, she underwent an Automatic Implantable Cardioverter Defibrillator (AICD) implantation with Medtronic EVERA MRI S DR SureScan DDMC3D4 (SN: PHZ619818S) on 04/04/2019. She had severe pain and a raised pruritic lesion over the surgical site (hyperpigmented, hypertrophic plaque over the left shoulder in the implantation site which was managed conservatively. There were no further episodes of VT noted after the implantation. Endocrinology and Hematology consultations were continued for the patient.
Thalassemia is one of the most common hereditary diseases found mainly among Mediterranean countries, north-coast African countries, the Middle East, Southern China, India, and South America [6]. The complications due to ineffective erythropoiesis in thalassemia may be prevented by transfusion therapy. However, transfusion therapy results in iron overload and secondary hemochromatosis complications. Even the patients on intensive chelation therapy suffer from cardiomyopathy and arrhythmic symptoms at a certain point in time [7]. There is no native pathway for body iron elimination and gastrointestinal absorption may be increased due to the suppression of hepcidin [8,9]. An increase in the iron load results in the appearance of iron in the circulation and this iron enters different organs through different pathways other than those mediated by transferrin. Hence, chelation therapy is the only way to prevent the accumulation of iron in organs like the liver, heart, and endocrine organs. Though chelation therapy has improved the mortality rates, cardiomyopathy due to iron overload is the main cause of morbidity and mortality in patients with β-thalassemia major [4].
The physiological iron absorption by the heart through transferrin receptors is a regulated process that does not lead to iron overload. The low molecular weight Non-Transferrin-Bound Iron (NTBI) appears in circulation when the transferrin-binding capacity is increased. NTBI exhibits oxidative activity and enters the heart through non-specific cation channels that are poorly regulated, resulting in cardiac overload [7]. Iron overload also affects the conduction system of the heart, resulting in conduction delays and heart block [10]. The presence of iron levels in the myocardium appears to be associated with the occurrence of arrhythmias and conduction abnormalities. An increase in the labile iron concentration increases the oxidative stress in the myocyte. Conduction or repolarization disturbances, arrhythmias, and diastolic or systolic dysfunction may occur when the calcium, potassium, and sodium ion channels get disrupted. Abnormal electrocardiogram was reported in 46%, abnormal T waves in 34%, and right bundle-branch block was reported in patients with β thalassemia major [11].
In this case, the patient had iron overload despite undergoing chelation therapy. The RVOT VT was confirmed with HOLTER monitoring. There were progressive AV conduction abnormalities. Ventricular arrhythmias and atrial arrhythmias in patients with thalassemia are rare, and mostly resistant to antiarrhythmic therapy [12]. We tried to chemically cardiovert the patient but normal heart rhythm could not be achieved. Hence, she had to be electrically cardioverted. Given recurrent VT and progressive first-degree AV block, AICD implantation was done. Her intrinsic PR interval progressively increased from 300 msec to 420 msec. On a four-year follow-up, her left ventricular function remained stable. No further episodes of ventricular tachycardia on AICD interrogation. Ectopic subsided after chelation therapy. MRI to assess iron deposit in the heart was limited by artifacts due to ICD lead and pulse generator on the chest. However, an MRI of the liver showed a decrease in the iron load of the liver and the liver appears to show intrinsic dark signal intensity compared to adjacent muscle in T2 and liver iron concentration decreased from 4.83 mg/kg to 2.35 mg/kg.
To reduce the transfusion burden, she is planning to start on luspatercept injection. Gene therapy is not yet available outside US trials. Transplantation is deferred due to cardiac and liver iron overload and the complications associated with it.
Complications arising from ineffective erythropoiesis in thalassemia are managed with transfusion therapy. However, repeated blood transfusions may result in harmful iron accumulation in the endocrine glands and heart. Iron overload-induced cardiomyopathy is the primary cause of morbidity and mortality in patients with β-thalassemia. In this case report the patient suffered from recurrent episodes of RVOT VT due to iron toxicity which was effectively managed through AICD implantation. The key highlight of this case is that the patient has been free of RVOT VT for the past four years since the AICD implantation.
- Rund D, Rachmilewitz E. β-Thalassemia. New England Journal of Medicine. 2005;353:1135-1146. Available from: https://doi.org/10.1056/nejmra050436
- Ali S, Mumtaz S, Shakir HA, Khan M, Tahir HM, Mumtaz S, et al. Current status of beta-thalassemia and its treatment strategies. Mol Genet Genomic Med. 2021;9(12):e1788. Available from: https://doi.org/10.1002/mgg3.1788
- Lei M qing, Sun L Feng, Luo X Sheng, Yang X Yang, Yu F, Chen X Xia, et al. Distinguishing iron deficiency anemia from thalassemia by the red blood cell lifespan with a simple CO breath test: a pilot study. J Breath Res. 2019;13(2):026007. Available from: https://doi.org/10.1088/1752-7163/aafe4f
- Borgna-Pignatti C, Galanello R. Thalassemias and related disorders: quantitative disorders of hemoglobin synthesisatology. In: Wintrobe’s Clinical Hematology. 11th ed. Philadelphia: Lippincott Williams & Wilkins. 2004.
- Pippard MJ, Warner GT, Callender ST, Weatherall DJ. Iron Absorption And Loading In Β-Thalassæmia Intermedia. The Lancet. 1979;314(8147):819-821. Available from: https://doi.org/10.1016/s0140-6736(79)92175-5
- Weatherall DavidJ. The inherited diseases of hemoglobin are an emerging global health burden. Blood. 2010;115(22):4331-4336. Available from: https://doi.org/10.1182/blood-2010-01-251348
- Wood JC. Cardiac Complications in Thalassemia Major. Hemoglobin. 2009;33(sup1):S81–6. Available from:https://doi.org/10.3109/03630260903347526
- Papanikolaou G, Tzilianos M, Christakis JI, Bogdanos D, Tsimirika K, MacFarlane J, et al. Hepcidin in iron overload disorders. Blood. 2005;105(10):4103-4105. Available from: https://doi.org/10.1182/blood-2004-12-4844
- Wongjaikam S, Kumfu S, Chattipakorn SC, Fucharoen S, Chattipakorn N. Current and future treatment strategies for iron overload cardiomyopathy. Eur J Pharmacol. 2015;765:86-93. Available from: https://doi.org/10.1016/j.ejphar.2015.08.017
- Auger D, Pennell DJ. Cardiac complications in thalassemia major: Iron and the heart. Ann N Y Acad Sci. 2016;1368(1):56-64. Available from: https://doi.org/10.1111/nyas.13026
- Ramazzotti A, Pepe A, Positano V, Scattini B, Santarelli MF, Landini L, et al. Standardized T2* Map of a Normal Human Heart to Correct T2* Segmental Artefacts; Myocardial Iron Overload and Fibrosis in Thalassemia Intermedia Versus Thalassemia Major Patients and Electrocardiogram Changes in Thalassemia Major Patients. Hemoglobin. 2008;32(1–2):97–107. Available from: https://doi.org/10.1080/03630260701879514
- Bayar N, Arslan Ş, Erkal Z, Küçükseymen S. Sustained Ventricular Tachycardia in a Patient with Thalassemia Major: Sustained Ventricular Tachycardia. Ann Noninvasive Electrocardiol. 2014;19(2):193-197. Available from: https://doi.org/10.1111/anec.12085