
More Information
Submitted: June 27, 2025 | Approved: July 22, 2025 | Published: July 23, 2025
How to cite this article: Cheikh G, Diallo AD, Hanifa MI, Dioum M, Leye MCBO. Right Pulmonary Artery Arising from the Aorta: about an Observation. J Cardiol Cardiovasc Med. 2025; 10(4): 104-105. Available from:
https://dx.doi.org/10.29328/journal.jccm.1001216
DOI: 10.29328/journal.jccm.1001216
Copyright license: © 2025 Cheikh G, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.

Right Pulmonary Artery Arising from the Aorta: about an Observation
Gaye Cheikh1*, Diallo AD2, Hanifa MI2, Dioum M2 and Leye MCBO2
1Cardiology Department, National Hospital Center Cheikh Ahmadou Khadim Touba, Senegal
2Cardiology Department of National University Hospital Center, Fann, Dakar, Senegal
*Address for Correspondence: Gaye Cheikh, Cardiology Department, National Hospital Center Cheikh Ahmadou Khadim Touba, Senegal, Email: [email protected]
Hemitruncus is a rare congenital anomaly in which one pulmonary artery branch, usually the right, arises from the ascending aorta just above the aortic sinuses. In contrast, the main pulmonary artery and the other pulmonary branch arise in their normal position. First described in 1868 by Fraentzel [1], its prognosis depends on timely surgical management, given the risk of Eisenmenger’s disease. Treatment is surgical and must be initiated early.
We report the case of a 19-month-old infant with hemituncus whose circumstance of discovery was dyspnea.
A 19-month-old male infant from a non-consanguineous marriage, the 4th of 4 siblings with one case of abortion in the family. The pregnancy was carried to term, with no notion of resuscitation at birth. The symptoms reported by her mother began at 2 months of age: the mother noted a hyperpulsatility of the precordium associated with a staturo-ponderal delay. This necessitated several unsuccessful consultations, and with the onset of diaphoresis and dyspnea during feedings, he was referred to our department. His saturation was 93% on room air, his heart rate was 120 beats per minute, and his blood pressure was 110/70 mmHg. He had delayed weight-bearing and delayed reflex acquisition. Cardiac auscultation was normal. The patient was in congestive heart failure at the first consultation, with discrete labial cyanosis without digital hypoxia.
The electrocardiogram showed bi-atrial and left ventricular hypertrophy. On frontal telethorax, cardiomegaly with a supra-diaphragmatic peak, a right inferior arch overhang, and peri-hilar vascular overload, predominantly on the right. Cardiac ultrasound revealed a situs solitus heart with levocardia. The aorta normally emerges from the left ventricle, without obstruction. After a few centimeters, it gave way to a right pulmonary artery that filled correctly in systole without obstruction (Figures 1,2). The left pulmonary artery arises directly from the right ventricle. All cardiac cavities are dilated—suprasystemic pulmonary hypertension with ostium secundum-type atrial septal defect with right-to-left shunt. Cardiac angioscan confirmed the diagnosis, showing a 13 mm right pulmonary artery arising from the aorta. The left pulmonary artery arises directly from the right ventricle and measures 9.73 mm, with a functional pulmonary valve (Figures 3,4). Biological tests revealed microcytic hypochromic anemia at 9.3 g/dl. Medical treatment was based on furosemide, captopril, and spironolactone. A surgical cure after catheterization with reimplantation of the right pulmonary artery was indicated. The child died before surgery.
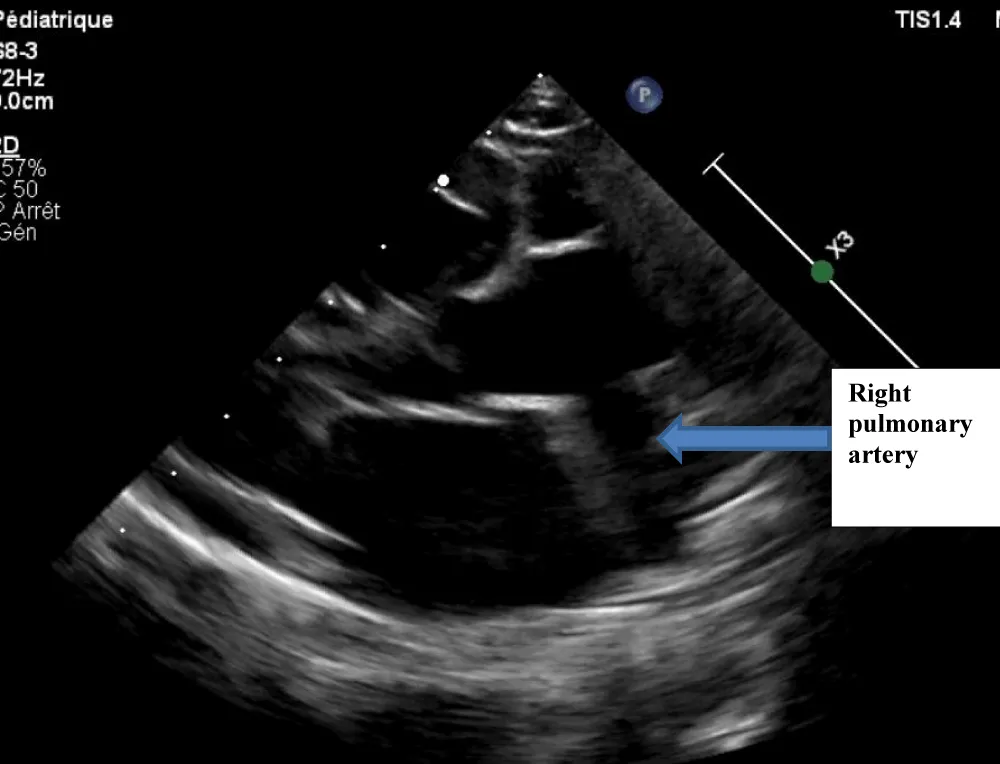
Figure 1: Echocardiography image showing the Right Pulmonary Artery (RPA) exiting the aorta.
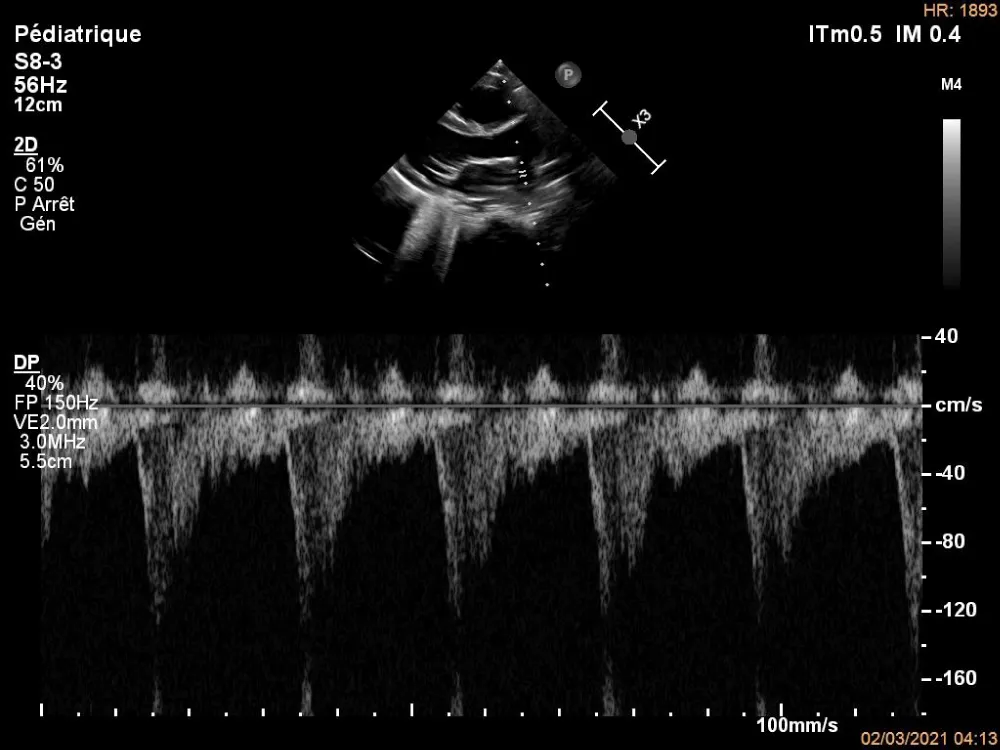
Figure 2: Pulsed Doppler echocardiography on the right pulmonary artery.
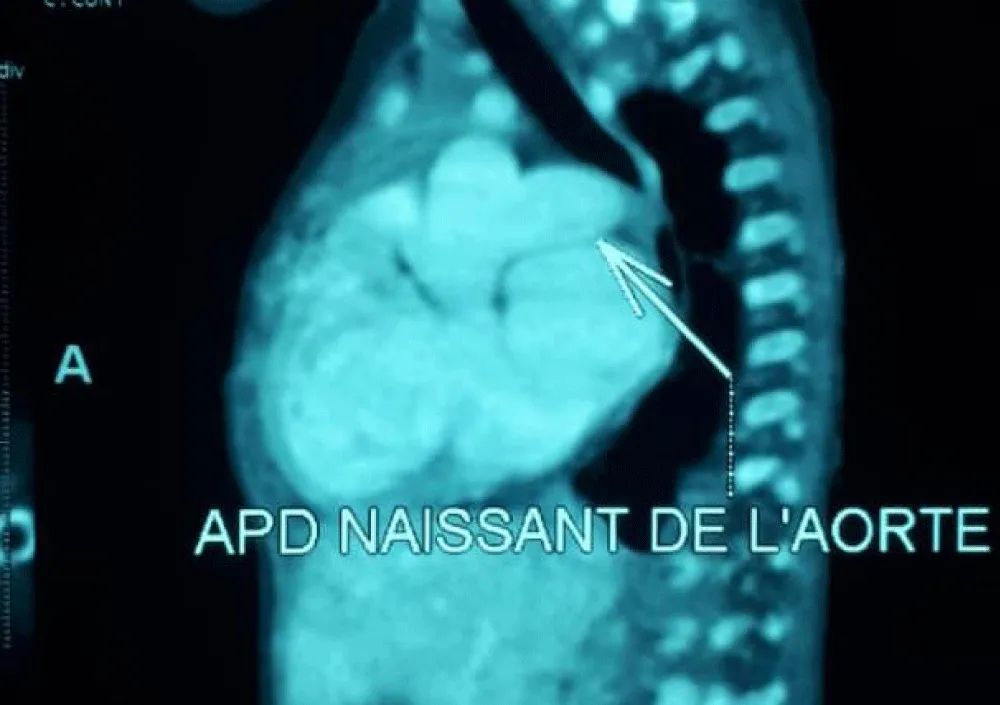
Figure 3: Right pulmonary artery arising from the aorta on cardiac angioscan./p>
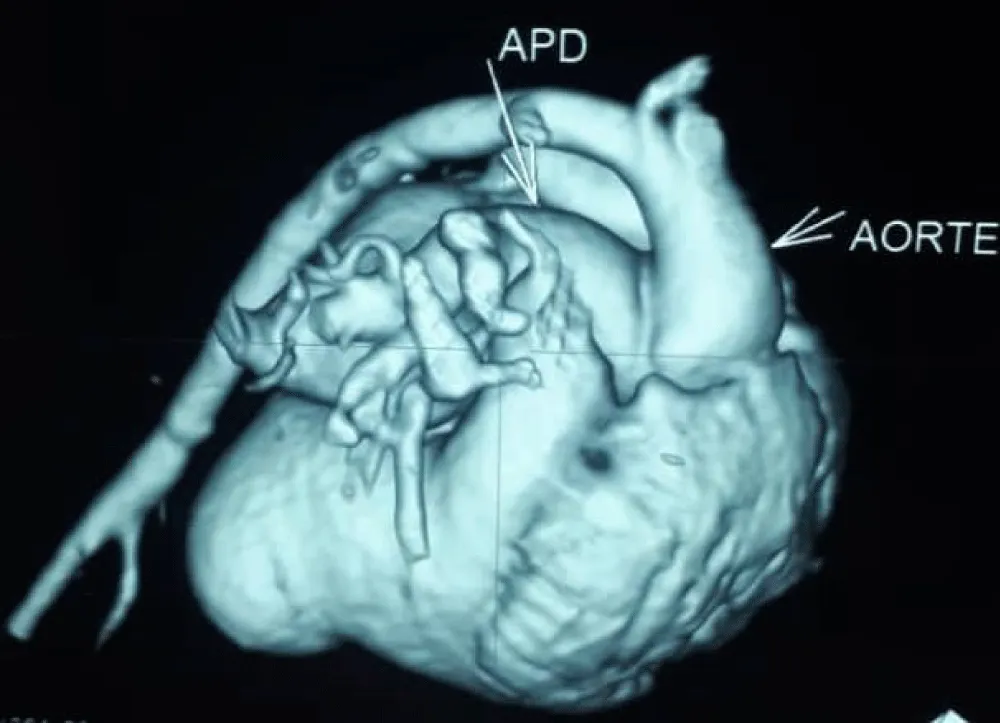
Figure 4: Cardiac angioscan reconstruction image showing right pulmonary artery exiting the aorta.
Hemitruncus is a very rare congenital cardiac anomaly. Edward Peng and all [2] found 9 cases over 29 years. The mean age at diagnosis was 14 days [3]. In a prospective study in the Hospital Infantil of Mexico “Federico Gomez” from 1991 to 2002, 12 patients were found with a mean age at diagnosis of 2 months. The circumstances of discovery are most often heart failure, as demonstrated by the case report by Edward W.K. Peng and all, who found 100% heart failure with only one case of cyanosis, or 11% of cases [2]. Echocardiography evokes the diagnosis in most cases, with confirmation by angiography or cardiac angioscanner. Edward and all reported echocardiographic diagnosis in 78% of cases [2]. According to Reyes and all [4], echocardiography evoked the diagnosis of hemitruncus in all patients, and this was confirmed by angiography in 8 patients, 66.6% of cases. Hemitruncus was isolated in our child. However, in the literature, it can be found associated with tetralogy of Fallot, aortic arch anomalies, or in the context of Di George syndrome [5,6].
Arterial hypertension is almost constant and rapidly severe in hemitruncus arteriosus. Pulmonary hypertension is present in 30% of cases by the 3rd month and in 70% of cases by the 12th month [3] and rapidly progresses to pulmonary vascular disease, with death in severe cases [3]. Riyadh M, et al. [5], with 36 years’ experience at the University of Toronto in Canada, found arterial hypertension in all their patients, with 4 cases of systemic or supra-systemic pulmonary hypertension out of the 16 patients included in the study.
However, early surgical management can prevent pulmonary vascular disease and death [3]. It should be performed before 6 months of age to avoid Eisenmenger’s syndrome. In our infant, the severity of the disease, late diagnosis, and suprasystemic pulmonary hypertension resulted in death.
Hemitruncus is a very rare congenital pathology with a very high morbi-mortality. Surgical cure remains urgent to avoid pulmonary arterial vascular disease.
- Fraentzel O Fall, Von E. Abnormal communication of the aorta and pulmonary artery. Arch Pathol Anat 1868;43:420–6.
- Peng EW, Shanmugam G, Macarthur KJ, Pollock JC. Ascending aortic origin of a branch pulmonary artery--surgical management and long-term outcome. Eur J Cardiothorac Surg. 2004;26(4):762-6. Available from: https://doi.org/10.1016/j.ejcts.2004.07.007
- Jayan J P P, Vijayalakshmi IB, Narasimhan C. A rare anomaly: 'hemitruncus'. Heart. 2011;97(12):1027. Available from: https://doi.org/10.1136/hrt.2011.222315
- Reyes de la Cruz L, Vizcaíno Alarcón A, Arévalo Salas A, Espinosa Islas G, Bolio Cerdán A, Arteaga Martínez M. Echocardiographic diagnosis of anomalous origin of one pulmonary artery from the ascending aorta. Arch Cardiol Mex. 2003;73(2):115-23. Available from: https://pubmed.ncbi.nlm.nih.gov/12894488/
- Abu-Sulaiman RM, Hashmi A, McCrindle BW, Williams WG, Freedom RM. Anomalous origin of one pulmonary artery from the ascending aorta: 36 years' experience from one centre. Cardiol Young. 1998;8(4):449-54. Available from: https://doi.org/10.1017/s1047951100007101
- Dodo H, Alejos JC, Perloff JK, Laks H, Drinkwater DC, Williams RG. Anomalous origin of the left main pulmonary artery from the ascending aorta associated with DiGeorge syndrome. Am J Cardiol. 1995;75(17):1294-5. Available from: https://doi.org/10.1016/s0002-9149(99)80788-7